week 11: principal components & dimensionality reduction
Distress not yourself if you cannot at first understand the deeper mysteries of Spaceland. By degrees they will dawn upon you.
Edwin A. Abbott, Flatland: A Romance of Many Dimensions
goals this week
Principal components analysis (PCA) is a classic tool for data analysis. It is frequently used for data visualization in bioinformatics, but was developed first in physics and statistics. It can be traced back to a 1901 paper by Karl Pearson, where he discusses the "line of best fit."
The main reason we use PCA in data analysis is to reduce the dimensionality of our data.
what is dimensionality?
Suppose we have an \(n \times p\) data table where we have measured \(p\) variables (or features) in columns, for \(n\) observations in rows. For example, we might have single-cell RNA-seq data for \(n\) individual cells, measuring mapped read counts for \(p\) different genes in each cell.
We can imagine each observation (each cell) as a point in an \(p\)-dimensional Euclidean space. For example, in homework 09 we did K-means clustering of single cell RNA-seq data in two dimensions, for two genes kilA and defA.
In real RNA-seq data, the number of genes \(p\) is much larger than 2. Many other sorts of biological experiments these days generate "high-dimensional" data too. As soon as \(p\) gets bigger than 2 or 3, the dimensionality of the "space" that we're plotting our points (observations) in becomes difficult for us to visualize, and it's hard to see how points are clustered or correlated.
Here's something to watch out for. Maybe I'm interested in how cells are related to each other, in gene expression "space". Alternatively, I could also be interested in how genes are related to each other, in cell space. When you look at someone's PCA plot of their RNA-seq data, one first thing to think about is, what are the points - genes, or cells? To build on a consistent intuitive picture throughout these notes, I'm always going to talk about the observations (points) being cells, but we could just as easily transpose the analysis problem and talk about the genes as the points and the samples as the dimensions (features), in order to look at how genes cluster in the \(p\)-dimensional space defined by expression values in different samples.
There are a variety of techniques for reducing the dimensionality of high-dimensional data, so it can be visualized and studied more easily. PCA is one of the most commonly used.
a first glimpse at PCA
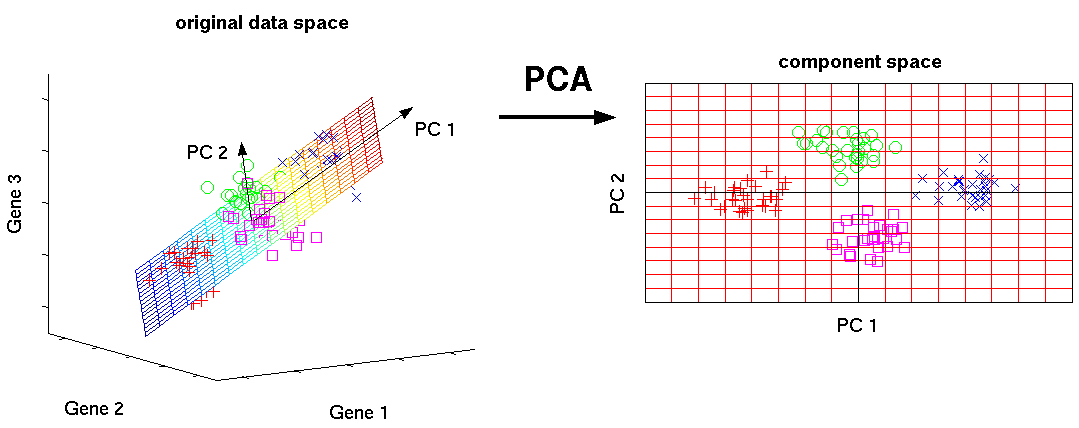 Figure from [Scholz 2006].
PCA aims to find projections of the data onto a new set of axes that best reveal underlying structure.
Figure from [Scholz 2006].
PCA aims to find projections of the data onto a new set of axes that best reveal underlying structure.
PCA aims to find a rigid rotation of the original \(p\) axes of your data, to a new set of \(p\) orthogonal axes. The first new axis is the vector that captures the highest variance in your data, projected along that vector. The second axis is the orthogonal vector that captures the next highest variance, and so on.
These axes are often called principal components (PCs). Calling the axes principal components is somewhat incorrect, but never mind. If we're nitpicky, principal components are actually supposed to be your data projected along each new axis, where the orthonormal unit axes themselves are called principal directions or, as we'll see, eigenvectors. Each PC captures a fraction of the overall variance in your data. The sum of these fractional variances equals the total variance in your data.
If you have fewer observations than dimensions (\(n < p\)), then only the first \(n\) principal components will capture nonzero variance, for the same reason that you can always perfectly fit a 2D line to two points.
Some applications of PCA in biological data analysis include:
-
determining the intrinsic dimensionality of a system [see Stephens et al. (2008) for an interesting example]. How many underlying features are generating the observed data? While we might collect data from a high number of variables, it's possible that there are a small number of underlying processes generating the data. PCA is good at finding these underlying processes when they are independent.
-
denoising data [see Alter et al. (2000) for an example]. Measurements are often confounded by experimental artifacts that can obscure the types of signals that we care about. By emphasizing features of the data that show patterns across observations and variables, PCA can pick out real underlying structure and remove underlying, uncorrelated noise.
-
visualizing clusters. In the k-means pset in week 09, we looked for clusters in gene expression patterns based on two genes, where it was easy to directly visualize the distributions of interest. But how can we see clusters when considering thousands of genes? Let's say, in a simple case, there are only two genes out of thousand that are differentially expressed between clusters. If we can find these two genes and scatter plot them, then we might be able to see our clusters. But how can we find them?
line of best fit
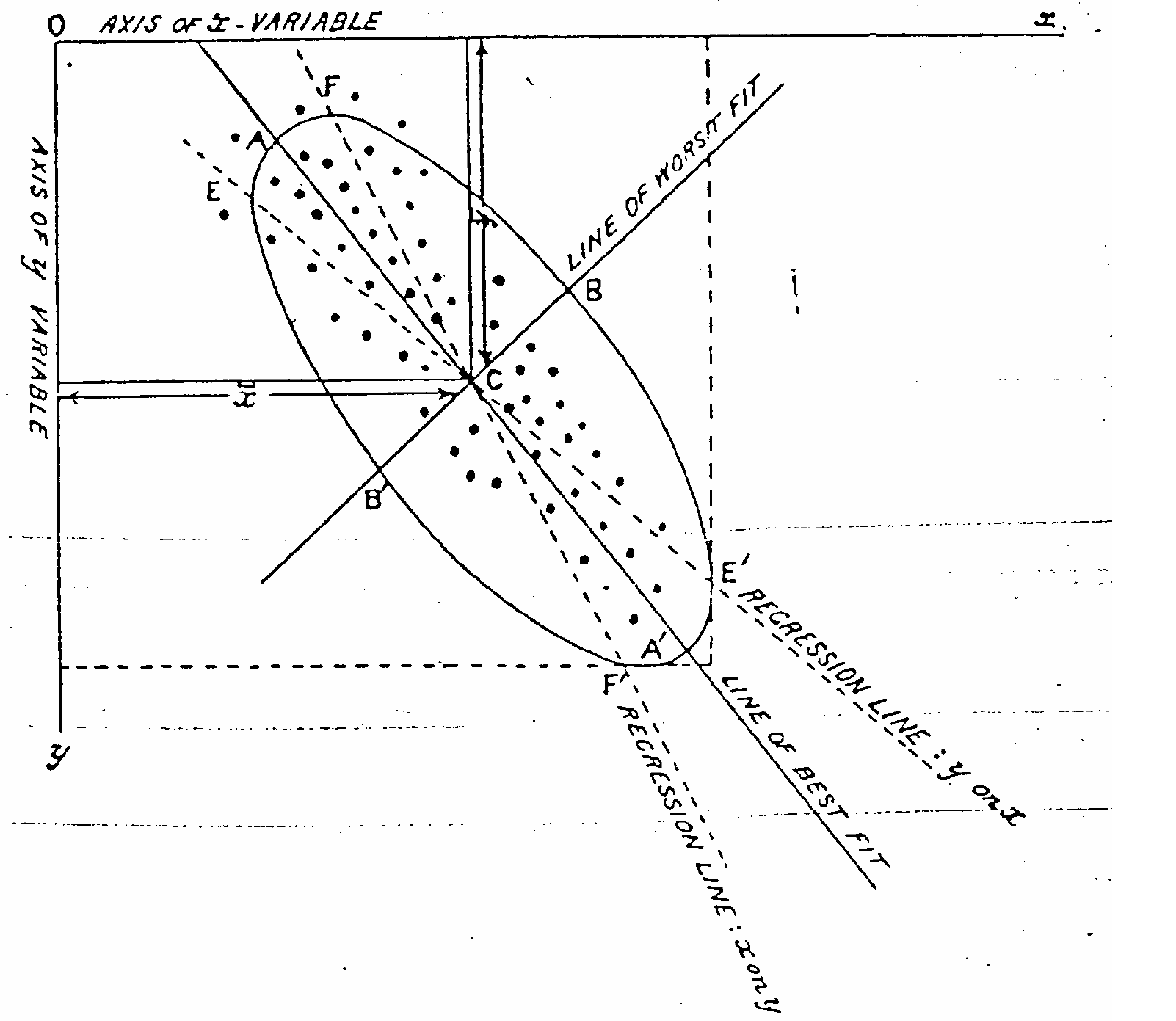 Figure from [Pearson 1901].
On lines and planes of closest fit to systems of points in space.
Figure from [Pearson 1901].
On lines and planes of closest fit to systems of points in space.
A place to start thinking about PCA is in two dimensions, with what Pearson called "the line of best fit" in a 1901 paper.
When we talked about regression, we talked about having an independent variable \(x\) and a dependent variable \(y\), such that our observation of \(y\) was a function of \(x\) plus some (typically Gaussian) noise: \(y = ax + N(0, \sigma^2)\), for linear regresssion. If we switched the dependent and independent variable, now looking to fit \(x = by + N(0, \sigma^2)\), in general we won't get the same regression line.
Pearson recognized that in many cases it isn't clearcut which variable is dependent and which is independent. It could be that they're really both noisy measurements, both dependent on some underlying cause, so they covary.
As Pearson put it with a concrete example, "the most probable stature of a man with a given leg length \(l\) being \(s\), the most probable length of leg for a man of stature \(s\) will not be \(l\)." The regression fits are not invertible. Pearson proposed that the "line of best fit" is one without bias towards either variable. But what residuals do we minimize?
We can find the line that minimizes the orthogonal distance between each point and the "line of best fit", which is tantamount to an assumption of Gaussian noise added to the orthogonal directions around an underlying vector.
That is, the "line of best fit" is equivalent to finding a line such that when the data points are projected onto it, the variance along that line is maximal. (Maximizing the variance along the line means minimizing the variance in all the other directions.) Pearson's line of best fit is the first principal component of a 2D data set. The remaining principal component is forced to be orthogonal to it, so its direction is completely determined by the best fit line.
Informally (we haven't defined "covariation" mathematically yet) we can see that once we've found the line of best fit that maximizes the variation it captures, there isn't any covariation left between this axis and the other axis (or axes, in more then two dimensions). If there were remaining covariation with some other axis, there would still be some variation we could capture by rotating our line of best fit. This idea holds up recursively in multidimensions. Having found the first principal component (with zero covariation with any other orthogonal axis), we rotate the system again in the remaining degrees of freedom and identify a second principal component, and so on.
a generative model: the multidimensional Gaussian
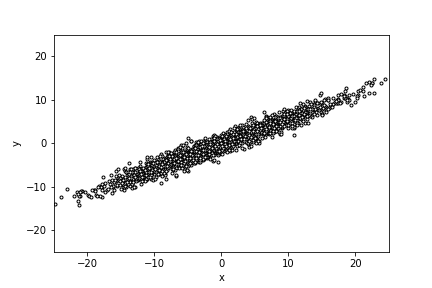 2000 data points sampled from a multidimensional Gaussian, along a 30 degree line
of best fit with a variance of 10 along the line, and 1 orthogonal
to it.
2000 data points sampled from a multidimensional Gaussian, along a 30 degree line
of best fit with a variance of 10 along the line, and 1 orthogonal
to it.
Suppose we wanted to generate sample data from a multidimensional Gaussian distribution that's tilted along some given line of best fit, with independent variances in each orthogonal direction in this tilted reference frame.
If the data cloud weren't tilted, this would be easy. If the variances were already independent in each normal-space axis in the data, then we'd just sample an independent Gaussian r.v. in each dimension, with the appropriate variance for that dimension.
We've already seen that I can sample a standard Gaussian random variable \(Z\) with mean 0 and std. dev. 1, then scale it to any new std. dev. in one dimension (i.e. just a scalar). In multiple dimensions, I just do this in each dimension independently. I sample a random variable \(Z_j\) independently in each dimension \(j\) to generate a sphere, then stretch that sphere by multiplying each axis by that dimension's standard deviation. Now I have an ellipse, stretched according to the standard deviation in each independent axis.
So now I want to imagine that this ellipse is in my bestfit-space coordinates (where the variance in each axis is independent), and I want to rotate into normal-space coordinates (x and y coords, in my 2D example.) The tilted "bestfit-frame" is defined by orthonormal unit vectors: the line of best fit, and the vector orthogonal to it. If you tell me the bestfit frame is rotated up by \(\theta\) degrees relative to the original x,y coordinate system, then one of these new vectors is \([ \cos \theta, \sin \theta ]\) (that's what we defined as "the line of best fit"), and the other is \([ -\sin \theta, \cos \theta]\) (that's the remaining orthogonal vector). Let's define a matrix \(\mathbf{W}\) whose columns are those vectors:
An important property of orthonormal matrices is that their transpose is their inverse: \(\mathbf{W}^T = \mathbf{W}^{-1}\). Therefore \(\mathbf{W}\mathbf{W}^T = \mathbf{I}\), where \(\mathbf{I}\) is the identity matrix (diagonal entries 1, all others 0).
If I have an \(n \times 2\) matrix \(\mathbf{X}\) of \(n\) data points in normal-space 2D (x,y) coords, then to rotate those points into bestfit-space coords \(\mathbf{Y}\), I would do \(\mathbf{Y} = \mathbf{X} \mathbf{W}\). The matrix multiplication is taking the dot product of each of my points \(\mathbf{x}_i\) with each of the bestfit-space vectors \(\mathbf{w}_p\): calculating the projection of \(\mathbf{x}_i\) onto the \(\mathbf{x}_p\) axis. Conversely, if I have an \(n \times 2\) matrix \(\mathbf{Y}\) of data points in bestfit-space (\(\mathbf{w}_1, \mathbf{w}_2\)) coords, I can rotate those coords back into normal-space coords by \(\mathbf{X} = \mathbf{Y} \mathbf{W}^T\).
This idea of rigidly rotating the original ("ambient") coordinates into and out of bestfit ("pca") coordinates generalizes from 2D to any number of dimensions \(p\).
Putting these ideas together, I can sample an \(n \times p\) matrix \(\mathbf{X}\) of \(n\) points in a \(p\)-dimensional space from a multidimensional Gaussian by:
where:
-
\(\mathbf{Z}\) are your independently sampled standard Gaussian random variables, with mean 0 and s.d. 1;
-
\(\mathbf{\Lambda}^{\frac{1}{2}}\) is just a diagonal matrix with elements that are the standard deviations \(\sigma_j\) for each of the \(p\) axes in bestfit-space. They're ordered from largest to smallest, because the first dimension \(\mathbf{w}_1\) is the "line of best fit". The reason for the obscure notation \(\mathbf{\Lambda}^{\frac{1}{2}}\) will become apparent.
-
The columns of \(\mathbf{W}\) are the orthonormal unit vectors that define our rotated "bestfit-space". We'll call these our eigenvectors.
For example, here's Python code to sample 2000 points from a two-dimensional Gaussian along a best fit line of \(30^o\):
import numpy as np
n = 2000 # number of points
p = 2 # number of dimensions
theta = 30./360. * 2. * np.pi # 30 degrees, in radians
# Define the unit eigenvectors of our rotated "bestfit space"
W = np.array( [ [ np.cos(theta), -np.sin(theta) ],
[ np.sin(theta), np.cos(theta) ]])
# Sample Z in bestfit-space
Z = np.random.normal(size=(n,p))
# Stretch Z by the standard deviation in each independent direction; largest to smallest.
S = np.diag( np.array( [ 10.0, 1.0 ] ) )
Z = Z@S
# Rotate into normal-space coords
X = Z @ W.T
I used this code to generate the figure. You can use it (or code like it) to create a dataset where you can play with PCA where you know the correct answer (the correct eigenvectors that define "bestfit-space").
covariance
We jumped ahead of ourselves a bit. (I mean, just look at that scary word 'eigenvector'.) Let's now come at it again, from a more textbook direction.
Suppose we're given a bunch of data points from a multidimensional Gaussian; how would we describe that distribution? That is, what sufficient statistics suffice to parameterize it? Let's formalize the definition of covariance.
Recall that the sample variance, \(s^2\), was used to describe the spread of a set of \(M\) observations of one variable with mean \(\bar{X}\) - the average of the squared residuals:
Recall that the sample variance \(s^2\) is usually calculated using \(\frac{1}{n-1}\) instead of \(\frac{1}{n}\): "Bessel's correction".
When we start to work with data points in more than 1 dimension (more than one variable per observation, in tidy-data-speak; i.e. more than one gene measured per sample or cell, in RNA-seq), it's possible that, in addition to the variance intrinsic to each of the single variables, there is covariation between the variables (genes). Covariation means that the two variables are correlated with each other.
The covariance calculation looks very similar to the variance calculation. In two dimensions, with a set of \(n\) observations of variables \(X\) and \(Y\) with individual means \(\bar{X}\) and \(\bar{Y}\), respectively - the average product of the residuals:
If X and Y tend to go up and down together, this'll give us a positive residual times a positive residual or a negative times a negative, and we'll get a positive covariance. If one goes up when the other goes down, we'll get a negative covariance - they're anticorrelated. If X and Y are uncorrelated, we'll get just as many pos x neg and neg x pos residuals as we get pos x pos and neg x neg, and we'll get a covariation that tends to zero.
With this definition in hand, we can rewrite the variance as the covariance of a variable with itself:
In p dimensions, we can compare all pairs of variables (dimensions, axes) and obtain a \(p \times p\) covariance matrix, denoted \(\Sigma\). For two variables \(X\) and \(Y\) the covariance matrix has four entries:
The diagonal entries are just the variances of \(X\) and \(Y\) themselves. The matrix is symmetric, which you can see by looking back at the equation for covariance. It's commutative - we're quantifying the same relationship when we talk about \(\mathrm{cov}(Y,X)\) as when we talk about \(\mathrm{cov}(X,Y)\).
When we generalize this matrix to \(p\) dimensions (variables), the notation will be easier if we index them \(1..j..p\), instead of talking about \(X\) and \(Y\) coords. These are indices of columns of our \(n \times p\) data matrix \(\mathbf{X}\). Thus \(x_{ij}\) is the \(i\)'th observation, \(j\)'th variable/dimension; each observation \(\mathbf{x}_i\) has \(p\) dimensions.) Each entry in the covariance matrix \(\Sigma_{jk}\) is \(\mathrm{cov}(j,k)\), the covariance between variables \(j\) and \(k\):
A multidimensional Gaussian (centered at the origin) is defined by a \(p\)-dimensional covariance matrix \(\mathbf{\Sigma}\).
If someone gives us a multidimensional Gaussian defined by \(\mathbf{\Sigma}\), how would we sample from it? Somehow we need to get from the covariance matrix to a rotated basis in which all covariances are zero, so we can deal just with independent variances along each of a set of orthogonal basis axes. Perhaps surprisingly, such a rotated basis always exists! Linear algebra tells us that we can use eigendecomposition to factor a covariance matrix (a covariance matrix is symmetric and positive semidefinite, in the lingo):
where \(\mathbf{\Lambda}\) is a diagonal matrix of the eigenvalues of \(\mathbf{\Sigma}\), i.e.,
and \(\mathbf{W}\) is a \(p \times p\) matrix whose columns correspond to each eigenvector, \(\mathbf{w}_{1 ... n}\).
I haven't proven here why an eigendecomposition finds a rotation with zero covariances - and I also haven't proven that this \(\mathbf{W}\) is the same as the \(\mathbf{W}\) we used to generate the data above, but it is. We're not a linear algebra class! But these things are true, and they're the ideas that are most important to hold on to.
Here's some other important facts:
-
The eigenvalues are the independent variances along each eigenvector; and their square roots are standard deviations. (Now you see why we had the notation \(\mathbf{\Lambda}^{\frac{1}{2}}\) when we generated a multidimensional Gaussian by stretching a standard Gaussian sphere by each dimension's standard deviation.)
-
The sum of the eigenvalues (the trace of matrix \(\mathbf{\Lambda}\)) is the total variance. This is equal to the total variance of the original data (the trace of the covariance matrix \(\mathbf{\Sigma}\)).
-
The eigenvector with the highest eigenvalue is the one that captures the most variance: it is the first principal component (PC1). The next highest eigenvalue defines PC2, and so on. The proportion of variance captured by a component is its eigenvalue divided by the total sum of the eigenvalues.
doing it in Python
Well, ok, I suppose the easy way to do it in Python is to call a canned PCA routine like sklearn.decomposition.PCA from the scikit learn machine learning package, but where's the fun in that? Is that how we do things around here?
One way to do the eigendecomposition is the way I just outlined above: calculate the covariance matrix \(\Sigma\), then do an eigendecomposition on it. With lower-level numpy calls, that'd look like:
# Given data array X, an (n,p) numpy array of n rows for our
# observations and p columns for our dimensions:
#
Sigma = np.cov(X, rowvar=False) # rowvar=False because numpy.cov() defaults to comparing rows not columns
Lambda, W = np.linalg.eigh(Sigma) # eigh() suffices because we know covariation matrix is symmetric
In practice it's more common and often more efficient to use a different decomposition technique called singular value decomposition (SVD). In biological data, we are often in the case where we have many more variables \(p\) than we have observations \(n\). For example, in the pset this week, we have \(p=2001\) genes measured in \(n=200\) cells. The data \(\mathbf{X}\) aren't really living in a \(p\)-dimensional space; two vectors can't describe more than a two-dimensional space, and \(n\) vectors can't describe more than an \(n\)-dimensional space. When \(p > n\), when we do PCA to successively maximize the variance captured by a set of orthogonal axes, we will have perfectly fit the data by the time we get to \(n\) axes, leaving us with at least \(p-n\) dimensions that have zero variance. We only care about the axes that describe nonzero variance - i.e. the ones with nonzero eigenvalues - so why bother calculating the zero ones? The technical term for the dimensionality of the space spanned by the data matrix \(\mathbf{X}\) is its rank, \(r\): \(r \leq \mathrm{min}(n,p)\).
Singular value decomposition can be applied to any matrix, but when we use SVD to do PCA, we specifically must apply SVD to the \(n \times p\) centered data matrix, which we'll call \(\mathbf{X}^{*}\). Centering means translating the data from its actual centroid to a centroid with 0's in each dimension, by subtracting the column mean in each dimension: i.e. \(x^{*}_{ij} = x_{ij} - \mathbf{\bar{x}}_j\).
If you were trying to prove how the SVD approach relates to the covariance matrix, a key fact is that \(\mathrm{cov}(j,k) = \frac{ \mathbf{x}^{*}_j \cdot \mathbf{x}^{*}_k} {n-1}\): the covariance between dimensions \(j\) and \(k\) is the dot product of the centered columns divided by \(n-1\). Thus \(\mathbf{\Sigma} = \frac{\mathbf{X}^{*\top} \mathbf{X}^*} {n-1}\).
Given a centered data matrix \(\mathbf{X}^*\), singular value
decomposition (using np.linalg.svd(), for example) gives you three
matrices:
That is, SVD decomposes the \(n \times p\) centered data matrix \(\mathbf{X}^{*}\) into the product of an \(n \times r\) matrix \(\mathbf{U}\), a diagonal \(r \times r\) matrix \(\mathbf{S}\), and a \(r \times p\) matrix \(\mathbf{W}^\top\), where \(r\) is the rank of \(\mathbf{X}^{*}\).
The \(r \times p\) \(\mathbf{W}^\top\) matrix is the transpose of our eigenvectors. (That is, its rows are our eigenvectors). Transpose it to \(\mathbf{W}\), \(p \times r\), and each column is a \(p\)-dimensional eigenvector \(\mathbf{w}_j\), for a total of \(r\) eigenvectors with nonzero eigenvalues. By "our" eigenvectors, I specifically mean that we know they're the same as the eigenvectors of the covariance matrix. If you're a linear algebraist: the columns of \(\mathbf{W}\), called the right singular vectors of the matrix we decomposed, are the eigenvectors of \(\mathbf{X}^{*\top} \mathbf{X}^{*}\), which is why they are the same as the eigenvectors of our covariance matrix \(\mathbf{\Sigma}\), which is why SVD works as an alternative approach to decomposing the covariance matrix.
The diagonal \(r \times r\) matrix \(\mathbf{S}\) is directly related to the nonzero eigenvalues of \(\mathbf{\Sigma}\):
NumPy's SVD function is np.linalg.svd(). Part of the pset this week is to learn how to use it in doing PCA.
A little more technical detail helps to see the relationship between SVD and the eigendecomposition of the covariance matrix. The covariance matrix \(\mathbf{\Sigma}\) is \(\frac{\mathbf{X}^{*\top} \mathbf{X}^*} {n-1}\). Its eigendecomposition is \(\mathbf{\Sigma} = \mathbf{W} \Lambda \mathbf{W^\top}.\) SVD gave us a different decomposition \(\mathbf{X}^* = \mathbf{U} \mathbf{S} \mathbf{W^\top}\). So:
with me so far?
OK, so you've centered your data matrix and done SVD; now you've got your \(p \times r\) \(\mathbf{W}\) matrix with eigenvectors as its columns, and you've got a diagonal \(r \times r\) matrix of eigenvalues \(\mathbf{\Lambda}\). You can check a few things to be sure that you're on the right track, including:
-
the sum of the eigenvalues down the diagonal of \(\Lambda\), i.e. its trace \(\mathrm{tr}(\mathbf{\Lambda})\), is the total variance of your data, equal to the sum down the diagonal of your covariance matrix \(\mathrm{tr}(\mathbf{\Sigma})\).
-
the eigenvectors are orthogonal to each other: so the dot product of any pair \(j,k\) of them is \(\mathbf{w}_j \cdot \mathbf{w}_k = 0\).
-
the eigenvectors are unit vectors: they each have a length of 1, \(\lVert \mathbf{x}_j \rVert = 1\).
"scores": plotting data into lower-dimensional PC space
Now we can use our new orthonormal basis \(\mathbf{W}\) to rotate the data along successive "lines of best fit", rotating it into a new coordinate system where the data only has independent variances in each axis and no covariation. In the rotated PCA space, the vector coordinates for observation (row) \(\mathbf{x}_i\) in the new directions of each eigenvector \(\mathbf{w}_j\) are just the dot products between \(\mathbf{x}_i\) and each eigenvector (each column of \(W\)). To project all the centered data observations onto one eigenvector is a linear combination \(\mathbf{X}^* \mathbf{w}_j\). To project all the centered data observations onto all the eigenvectors is \(\mathbf{Y} = \mathbf{X}^* \mathbf{W}\).
That's matrix \(\mathbf{X}^*\) multiplied by vector \(\mathbf{w}_j\). Don't be confused by the \(^*\) on the centered data matrix \(\mathbf{X}^*\). It's our notation for the centered data matrix.
The rows of \(\mathbf{Y} = \mathbf{X}^* \mathbf{W}\) are the original data observations, re-centered on the origin, and rotated into a coordinate system with successive "lines of best fit" to capture independent variance components, from largest to smallest.
Each element \(Y_{ij}\) is a PC score: the score (location) for data point \(i\) in principal direction (axis) \(j\).
If we take column \(j\) of \(\mathbf{Y}\) (and let's call it \(\mathbf{Y}^j\)), this is an array of \(n\) PC scores (locations along this PC axis) for the \(n\) data points in PCA direction \(j\). By definition of how we did PCA, the variance of this \(\mathbf{Y}^j\) array of PC scores is \(\lambda_j\) (the eigenvalue for the \(j\)'th PC). This set of \(Y^j\) columns is called the principal components of the dataset.
To visualize the data in a low-dimensional plot, we just take the first \(q\) eigenvectors with the largest eigenvalues, instead of all \(r\) of them. That is, we take the \(q\) leading columns of \(W\) (assuming we have it in order of largest to smallest eigenvalue) to get a \(p \times q\) matrix we'll call \(\mathbf{W}_q\), then:
gives us a \(n \times q\) dimensional projection of our centered \(\mathbf{X}^*\) onto a \(q\)-dimensional PC subspace. Typically we'd have \(q=2\) and we'd plot our data in two dimensions, PC1 versus PC2.
Again, the fraction of variance captured by PC1 is \(\frac{\lambda_1}{\sum_j \lambda_j}\), and PC2 captures a fraction \(\frac{\lambda_2}{\sum_j \lambda_j}\).
"loadings": which variables are most important
The elements \(w_{jk}\) of each eigenvector \(\mathbf{w}_j\) are called the PC loadings. The eigenvector is a "line of best fit" in the decorrelated rotation of our data. The vector direction tells us what's important to this vector. Specifically, the largest elements \(w_{jk}\) are the most important variables that are contributing variance in this eigenvector direction.
So in addition to plotting the data observations in the low-dimensional PCA plot, it's also useful to plot the variables, by treating the rows of the \(p \times q\) matrix \(\mathbf{W}_q\) as points in \(q\) dimensions. Variables that aren't contributing much variance along a principal axis will fall near the origin. Variables that do contribute will fall away from the axis, and correlated or anticorrelated variables will lie along the same PC direction.
By looking at how PC scores for data observations (e.g. cells) cluster in PC space, you see clusters of related data points. This might tell you something about cell types in your data. By looking at how PC loadings for variables (e.g. genes) cluster in PC space, you see which genes are contributing most to each principal axis. This might tell you something about which genes are responsible for differentiating cell types.
You'll sometimes encounter a specific kind of PCA plot, called a biplot, that shows both scores and loadings on the same graph, usually with the scores (observations) as points and the loadings (variables) as vectors (arrows).
"reconstruction": denoising data
The singular value decomposition has the property that if, besides taking the leading \(q\) columns of \(\mathbf{W}\) to get \(\mathbf{W}_q\), we also take the diagonal \(q \times q\) matrix \(\mathbf{S}_q\) (the singular values in \(\mathbf{S}\) that correspond to the leading \(q\) eigenvalues), and the corresponding leading \(q\) columns of \(\mathbf{U}\) to get a \(n \times q\) matrix \(\mathbf{U}_q\), we can "reconstruct" the data by squeezing it down onto just the first \(q\) principal axes, then rotating it back into the original data frame of reference:
Then adding the centroid back removes the centering and translates us to reconstructed data \(\mathbf{X}_q\). This is still an \(n \times p\) matrix, same as the original data \(\mathbf{X}\), but we've removed all the variance due to all the other principal axes other than the most important \(q\) of them.
This can be useful for denoising data, when we're in a situation where we think the top principal axes are signal, and the rest are noise. We can be in a situation where the data are varying in a few directions for interesting reasons with large variance in those directions, but where there's so many other directions contributing small noise variances, the total noise overwhelms the signal.
Again a little more technical detail might help clarify. A linear algebraist can easily show that \(\mathbf{X}^* \mathbf{W} = \mathbf{U} \mathbf{S}\), so \(\mathbf{Y} = \mathbf{U} \mathbf{S}\) gives us an alternative way of calculating all principal components, and \(\mathbf{Y}_q = \mathbf{U}_q \mathbf{S}_q\) is an alternative way of just calculating the leading \(q\) of them. When we do \(\mathbf{X}^*_q = \mathbf{U}_q \mathbf{S}_q \mathbf{W}^{\top}_q\), we're taking the data \(\mathbf{Y}_q\) in PC coordinates, and using the transpose \(\mathbf{W}_q^\top\) to rotate it back into the original centered data coordinates.
how many components are signal, how many are noise?
How many of our eigenvectors we should be taking seriously? This is where we can analyze the eigenvalues, which are the variance along each principal component.
It's common to plot the eigenvalues, or to plot the cumulative sum of the normalized eigenvalues. The latter graph shows the proportion of variance explained by the first \(q\) principal axes. This can be used to motivate retaining some subset of components for analysis. For example, if the first four principal components describe ~100% of the variance, you can retain just the first four components without losing any sleep. Usually it's not so clear, but that's the idea.
Another strategy is to compare the eigenvalues from the real data set to the eigenvalues expected from a matrix containing only random noise (ideally on the same spectrum as the noise in the real data). Any components above what is expected by chance alone would be more likely to be signal components you'd want to take seriously.
PCA is primarily for visualization and exploration
To prove what PCA is doing, from the standpoint of a generative probability model, we started with a unimodal multidimensional Gaussian distribution. We can then prove that PCA finds a new rigid rotation of the data (a new orthonormal basis) in terms of orthogonal directions of independent variance, such that there is no covariance between axes.
When we apply PCA to some general dataset that might have multiple clusters (modes) and non-Gaussian distributed points, PCA will still find an orthonormal basis in terms of independent variances. But if the data aren't Gaussian, then we might want a "line of best fit" to mean something other than the line that minimizes squared orthogonal residuals, for much the same reasons that we saw cases where linear regression fails. This may especially be a problem when the data have fat-tailed noise - when there are significant outliers. If we're going to ascribe meaning to principal axes, by taking PC loadings seriously, we might be able to get more relevant principal axes by devising variants of PCA that don't assume Gaussian-distributed data. There seems to be a statistical cottage industry of such "robust PCA" methods.
But if all we're trying to do is visualize and explore our data, and we're just trying to throw down our data into 2D so we can look at it, what the heck, finding a projection into a couple of dimensions that has some notion of maximizing the spread of the data seems reasonable.
Another thing to keep in mind, though, is that PCA is based on an assumption of a unimodal Gaussian distribution -- it doesn't know or care that you're probably using it to look for clusters. Doing a rigid rotation of a unimodal multidimensional Gaussian makes sense. Doing a rigid rotation of an arbitrary multimodal data distribution may or may not make sense. For example, you might have two clear clusters of data in your original data space that end up completely superposed in your PCA plot, just because they're somewhat less separated (less variance in the direction that separates them) than the variance that's separating other clusters in your data. To get a more faithful representation of clusters in p-space packed down into 2-space, we might need to resort to some sort of nonlinear projection of our data. Two popular nonlinear projection techniques these days are t-SNE and UMAP.
further reading
Joliffe and Cadima, 2016 is an impressively brilliant and compact review of PCA. Not only does it explain PCA's strengths and weaknesses from a user perspective, it tersely describes its mathematical foundation, including a crisp explanation of how you obtain an eigensystem problem when you seek a unit vector direction that maximizes variance -- you write down the constrained optimization with a Lagrange multiplier, and the equation for an eigenvector pops out as the solution.
These notes were originally written by Tim Dunn for MCB112 in fall 2016, but have been substantially revised, as my own understanding of PCA slowly improved over the years -- now any errors are my fault, not Tim's.