pset 03: the adventure of the undead genes
Your lab has been doing RNA-seq experiments with phage T4 in a time course after infection to measure its gene expression program. These RNA-seq experiments also measure host mRNAs, but you haven't really been paying attention to host mRNA expression levels, because you know that the phage aggressively shuts off host transcription and rapidly degrades the host genome down to nucleotides.
Moriarty recently published a paper where he describes what he calls the "thanatotranscriptome" of the host bacterial cell. He claims that after a lethal global transcriptional inhibition, he sees that many host mRNAs are differentially upregulated, as if a gene expression program is playing out after death - even in the absence of new RNA transcription. How can this be?
To show this, Moriarty has done RNA-seq time course experiments where he uses a drug (rifampin) to inhibit RNA polymerase at time 0. By using rifampin inhibition, he is studying a simple system of global transcription inhibition and death, independent of any complexities of phage infection. Rifampin is a fast-acting bacterial RNA polymerase inhibitor. For the purposes of the pset, assume it works instantly, and on all genes: no new RNA synthesis after time 0.
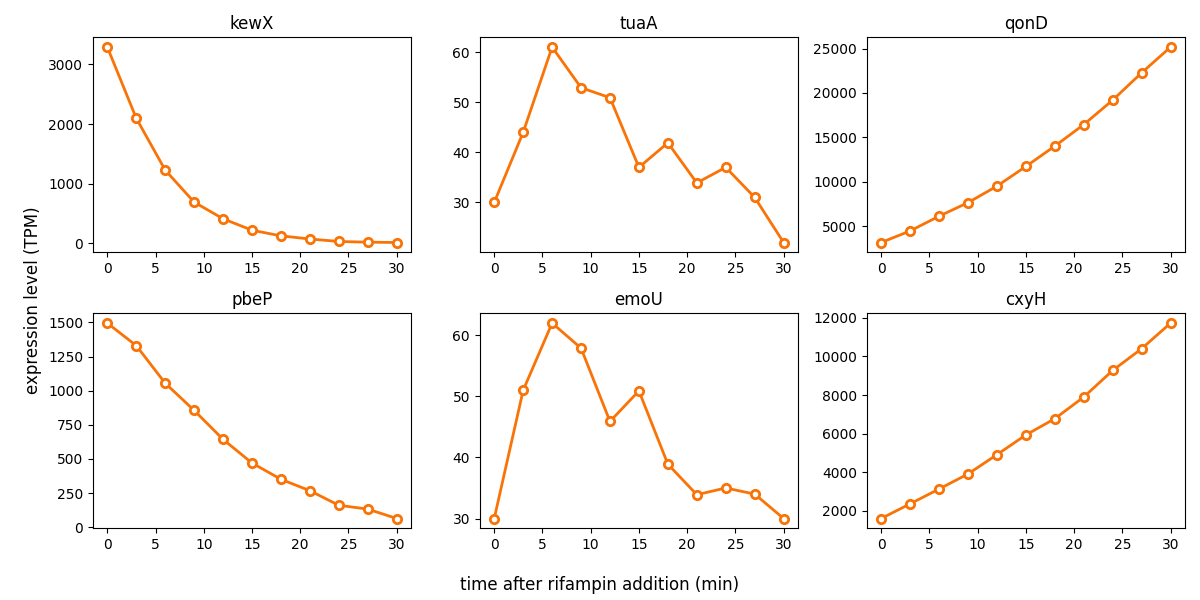
Some of his observations are shown in the figure below. He inhibits host transcription at time t=0 with rifampin treatment, then takes RNA-seq measurements in untreated cells (t=0) and at ten time points in 3 minute increments (3'-30') after the transcription block, and does RNA-seq on these 11 time points. He observes three broad classes of host genes. The two genes on the left (kewX and pbeP) are examples that go down exponentially, as one probably expects after transcription is shut off. The two genes in the middle (tuaA and emoU) go up, then down. The two genes on the right (qonD and cxyH) go steadily up over the time course. Moriarty calls kewX and pbeP "early" genes, tuaA and emoU "middle" genes, and qonD and cxyH "late" genes in his proposed post-death thanatotranscriptome expression program.
RNA can be produced not only by transcription from DNA using an RNA polymerase enzyme, but also from an RNA template by an RNA-dependent RNA polymerase. Many RNA viruses encode an RNA-dependent RNA polymerase that they use to replicate. I once forgot this fact in an interview for a PhD program and the interviewer called me the single stupidest prospective PhD student he had ever encountered. I met him again later when I became a faculty member at his university. RNA-dependent RNA replication has nothing to do with what's happening in Moriarty's data in the host bacterium. It seems really unlikely to you that any mRNA levels could be increasing after total inhibition of new RNA transcription, no matter how much Moriarty goes on about the possibility of an undiscovered program of RNA-RNA replication. You suspect an artifact of some kind instead. You look hard at how he did the RNA-seq experiment, and there doesn't seem to be any problem there. If there's a problem, it must be in his interpretation of his data.
the data
The expression data for the paper are available in his Supplementary Data Table 1: Moriarty_SuppTable1 as a tab-delimited tabular data file.
It happens that you've also just been reading another paper from Irene Adler's lab that seems relevant. Adler et al. carefully and systematically measured mRNA synthesis rates (in mRNA/min) and mRNA halflife (in minutes) for every gene in the same bacterial host. Her data are available in their Supplementary Data Table 2: Adler_SuppTable2, also a tab-delimited tabular data file.
Download both data files, which you'll need for your work below.
Read the two files into two Pandas DataFrames.
1. check that the gene names match
In principle, the 4400 gene names in the two data files ought to match, but (sigh) you know that all kinds of things can go wrong in practice: people use different names for the same gene, spell them with different capitalization or punctuation or added spaces, and so on.
Write code (using Pandas methods where possible) to compare the gene names in the two files. Find the names that appear in Moriarty_SuppTable1 but not Adler_SuppTable2, and vice versa, if any.
You already have reason to be suspicious of the Moriarty et al. data table because he mentions in his paper that he exported the supplementary methods tables from Microsoft Excel. You've run across people on social media discussing not just one but two terrifying papers about how Excel has systematically corrupted the supplementary data files in many published genomics papers.
Write code (using Pandas methods where possible) to fix any corrupted gene names in Moriarty's data, so the two lists of gene names match. Give an explanation for what happened in Moriarty's names.
There's another problem in the gene names, where it's Pandas doing the corruption! Pandas causes the same corruption in both files, so it's less easy to find. See if you can find it.
2. tidy the data
Moriarty's data is a "wide" tabular data set that is not in "tidy"
long format. Use Pandas melt to turn it into a new "tidy" data frame
in long format. Because Moriarty called his columns 3m, 6m, etc.
with the m for minutes as part of the column name, you'll also need
to change the variables in your newly melted time column so they're
integers, not strings, dropping those m's.
Hint: the tidy version will have the gene names as its index,
and three columns: keep the class column from Moriarty's data,
and the time course data are melted to two columns. I called
these columns time and tpm.
Some operations on the data are easier in tidy form; some will be easier in wide form.
3. explore the data
Write code (using Pandas methods as much as possible) to:
- output the five genes with the highest mRNA synthesis rate. (i.e. in Adler_SuppTable2)
- output the five genes with the longest mRNA halflife. (i.e. in Adler_SuppTable2)
- output the five genes that have the highest ratio of expression at t=30' vs. t=0 (i.e. in Moriarty_SuppTable1)
(You might want to explore the data in other ways too, on your own, as you're doing the next part.)
4. merge the data
Write code to integrate Moriarty's data with Adler's, in a single Pandas data frame.
There are at least two ways to do this. One is to merge Moriarty's
original wide form data with Adler's so that you append Adler's two
columns onto Moriarty's table: now for each gene, you have Moriarty's
classification ('early', 'middle', 'late'), his TPM measurements at 11
time points, and Adler's synthesis and halflife data. The second way
is to create a new data frame that integrates only the data you need,
perhaps with some derived data columns of your own: for example,
one useful integration might be a table with columns class,
synth_rate, halflife, t30/t0 ratio, where you combine
Moriarty's classification, Adler's rates, and one or more
calculations that seem useful from your data exploration above.
To do the integration/merge of the two data tables, you can use either Pandas
concat
or
merge
methods.
5. figure out the explanation
Using the merged data frame, and the original frames too as needed, explore the data, however you want, making whatever plots you want to illustrate points you think are important.
What do you think is a more plausible biochemical explanation for why some genes appear to be upregulated in Moriarty's RNA-seq experiments?
allowed modules
NumPy, Matplotlib, and Pandas.
You can optionally use Seaborn for plotting, in addition to
Matplotlib. Seaborn provides a nice
stripplot
plot that's way preferable to box plots. I'm going to use it in my own
answer.
Set the %matplotlib inline "magic command" to
get inline static figures from matplotlib in a Jupyter Notebook page.
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns # optional
%matplotlib inline
turning in your work
Submit your Jupyter notebook page (a .ipynb file) to
the course Canvas page under the Assignments
tab.
Name your
file <LastName><FirstName>_<psetnumber>.ipynb; for example,
mine would be EddySean_03.ipynb.
We grade your psets as if they're lab reports. The quality of your text explanations of what you're doing (and why) are just as important as getting your code to work. We want to see you discuss both your analysis code and your biological interpretations.
Don't change the names of any of the input files you download, because the TFs (who are grading your notebook files) have those files in their working directory. Your notebook won't run if you change the input file names.
Remember to include a watermark command at the end of your notebook,
so we know what module versions you used, in case we have to
reproduce some bug exactly. For example,
%load_ext watermark
%watermark -v -m -p numpy,matplotlib,seaborn,pandas,jupyter