pset 04: the adventure of the erratic nucleotide
Your lab is using CRISPR spacer sequences to identify which SEA-PHAGE phages infect which bacterial hosts.
Bacterial CRISPR host defense systems work in part by obtaining small pieces of phage DNA (presumably from failed infections) and incorporating them into arrays in the bacterial genome. The bacterium transcribes these arrays, processes the transcripts into short RNAs. The short RNAs are then used to target CRISPR defense systems, recognizing complementary phage DNA or RNA sequences on an incoming phage, and destroying it somehow. (CRISPR systems have different ways of destroying the target phage or having the infected cell commit self-destruction.)
Therefore, the sequences that a bacterial genome has incorporated into
CRISPR arrays act like a memory of what phages have infected this
cell's ancestors. By finding complementary sequences in known phage
genomes, if we're lucky (if a similar phage is in the database) we can
infer what phage these were.
The idea is to sequence CRISPR spacers, then computationally match those short host sequences (\(\sim\)30-40nt) to SEA-PHAGE genome sequences. Matches suggest that this host was infected by this phage at some point.
Moriarty's erratic DNA sequencer
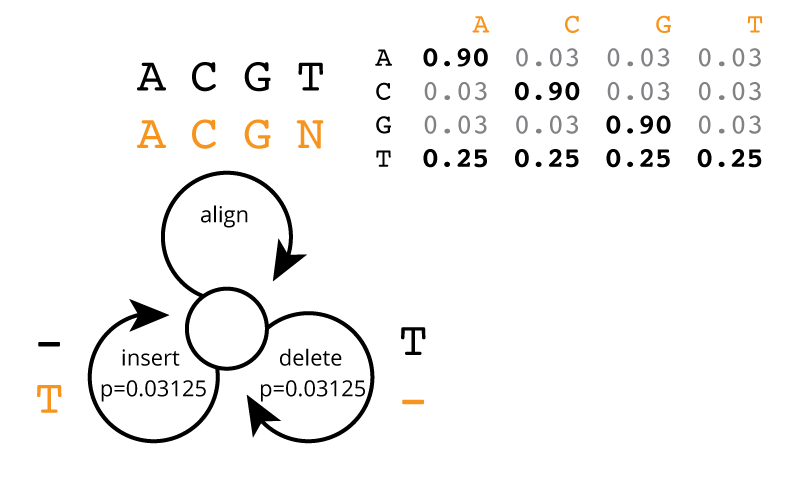
Moriarty is leading the project. He is using his latest invention, a new DNA sequencing machine that's faster and cheaper than any other existing technology. Unfortunately, his technology also has a peculiar error profile: it cannot sequence T's accurately. A, C, and G nucleotides are sequenced with 90% accuracy, but T positions are called uniformly randomly, 25% each base. Worse, T's are inserted (into the observed read) randomly 3.125% of the time (that's 1/32), and T's are deleted (from the actual sequence) 3.125% of the time.
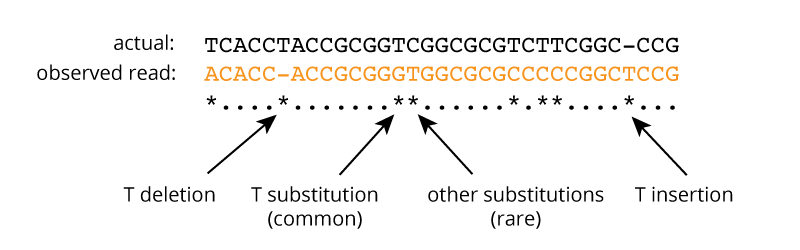
An example of a Moriarty read (in orange) aligned to the actual corresponding 32nt CRISPR spacer sequence is shown below. 8 asterisks mark the positions of basecalling errors: 1 T deletion, 1 T insertion, 5 T substitutions, and 1 substitution at another base (here, a C).
This error process is illustrated as a state machine in the figure below. The state machine consumes symbols from the actual sequence (black symbols) and generates symbols on the observed read. Each step of the machine, it follows one of the three arrows. At any step, it can insert a T with probability 1/32 (0.03125); at any T residue in the actual sequence, it can delete that T with probability 1/32; else, it generates a base aligned to the actual base, with probabilities as shown in the table: 90% accuracy for A|C|G, and uniformly random 25% for basecalling a T.
The state machine terminates when it tries to consume a residue on the actual sequence but there are none left. (Thus, it may still insert some T's at the end of the generated read sequence.)
Unfortunately, these basecalling errors seem to make Moriarty's reads useless. He has been unable to computationally map spacer reads to phage genomes, even using positive controls of known host-phage interactions and well-studied host CRISPR arrays. It's not hard to understand why it's failing. The expected identity of an ungapped alignment between a read and its source spacer (assuming uniform 25% each base) is only 74% (90% at 75% of the bases + 25% at 25% of the bases), and the insertions and deletions just make things worse. Standard read mapping methods assume at most one or two mismatches, and even homology search methods like BLAST and HMMER fail to resolve signal from noise on short sequence comparisons that are this divergent.
But here's where you come in. Even just a few weeks into MCB112, you know enough to make a customized alignment model, specific to Moriarty's error profile. You know that a custom alignment substitution matrix \(\sigma(b|a)\) can be derived as \(\log_2 \frac{p(b|a)}{f(b)}\), and thus (assuming uniform 25% base composition), you derive the alignment scores that should be used. Using what you've learned this week in MCB112, you sketch the following alignment score model at a white board, mirroring the generative probability model:
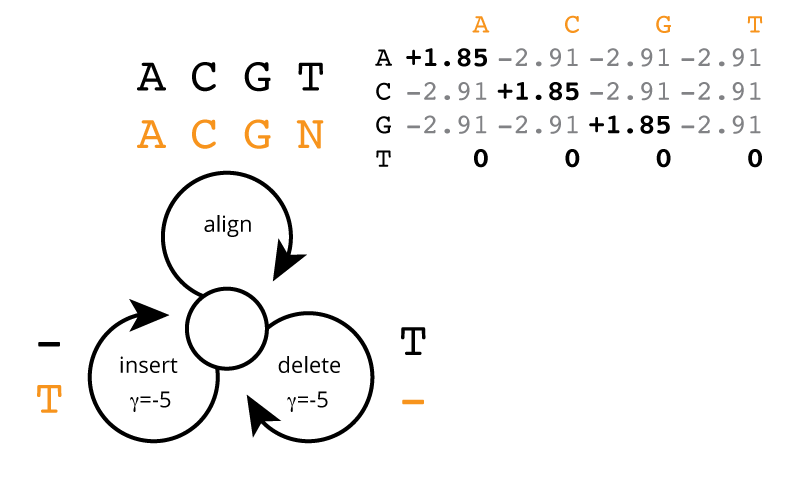
One thing you don't know from MCB112 yet is how to set the gap cost for insertion/deletion. You schedule a 60-second meeting with your professor, Holmes. Professor Holmes takes one glance at the insertion/deletion probability of 1/32 and says it's elementary: \(\log_2 \frac{1}{32}\) = -5, so your gap score should be -5. You have no idea how he did this, but whatever; you put in a -5 for the insertion/deletion score in the alignment model. You will verify (in section 2 below) that it's a reasonable choice. He also mutters something about missing a parameter if you were really doing this precisely right, but since we haven't seen hidden Markov models yet, he can't explain it to you.
1. implement this global alignment model
Hint: the optimal global alignment score for the sequences in the example figure above, for actual sequence TCACCTACCGCGGTCGGCGCGTCTTCGGCCCG and observed read ACACCACCGCGGGTGGCGCGCCCCCGGCTCCG, is 33.34 bits: 25 identical aligned A|C|G residues x 1.85 + 1 mismatch A|C|G alignment x -2.91 + 5 aligned T residues x 0 + 1 T deletion x -5 + 1 T insertion x -5 = 33.34. This gives you a known test case to try, and you can make others. I put the two sequences there so you can cut&paste them, since you can't do that from the figure.
Write a function to perform global sequence alignment between two sequences, the actual sequence and a read sequence, by dynamic programming, using the scoring model above.
Have your function take a gap score as an argument, because we're going to vary it in the next section.
Your function can also take a match score (for identical A|C|G aligned pairs) and a mismatch score (for mismatches involving A|C|G on the actual sequence), but we won't be varying them, so you can also just hardcode them if you prefer.
Have your function return the optimal global alignment score.
You do not need to find the optimal alignment itself (you don't need to implement the traceback phase of the dynamic programming, only the matrix fill phase).
2. check that the -5 gap score seems reasonable
You're not quite sure how Professor Holmes immediately stated that -5 was a good choice for gap score. Classically, gap scores are arbitrary parameters that have to be set empirically. One way to do this is to run a simulation where we score a bunch of positive control sequences, and a bunch of negative control sequences, and calculate the difference between mean positive and negative scores. We can then vary the gap score and optimize for the biggest gap, which should give us the best chance of discriminating positives from negatives.
Write a function that takes an actual sequence as input, and uses the probabilistic error model to generate a simulated read from it.
Use that function for generate positive control pairs, by making random (i.i.d., uniform base composition) 32nt "actual sequences" and sampling a read from each one.
Generate negative control pairs by generating two random i.i.d. 32nt sequences.
This is computationally expensive! My version took about a few minutes to run in total, over all the gap choices. Otherwise I would have done more than 2000 samples each, to sharpen up my estimates of these means. For each gap score choice in [ 0, -2, -4, -5, -6, -8, -10 ], collect scores for 2000 positive and 2000 negative simulated pairs; calculate the difference between the means.
Does -5 look like a reasonable choice? Why? Of course it does. Would Holmes lead you astray?
With only 2000 samples, there's a fair amount of noise in the empirically measured gap in bitscore between positive and negative controls; maybe about \(\pm 1\) bit. Don't be too concerned if the choice of -4 or -6 come out as the actual max for you. That's why I'm only saying "reasonable", not the clear maximum in this small simulation.
3. implement the alignment model for genome scanning
Hint: the recursion is the same. The only modifications you need are in initialization and termination steps of the DP algorithm. Now write a new dynamic programming function that modifies your global alignment routine to do global/local alignment: global with respect to the read, local with respect to the actual sequence. Using global/local ("glocal") alignment, you find matches anywhere in a long target genome sequence that you require to align to the entire query read sequence, not just part of it.
Have this function return the best score found in the target sequence, and the end position where that best score was found.
You don't need to find the start position of the best alignment, only the end. Finding the start as well would require a traceback phase to your dynamic programming, and I'm sparing you that.
4. demonstrate proof of principle
Moriarty doesn't believe your algorithm works, because he didn't think of it. He challenges you with an example read GCAGCGGAGGAACCAGACATACTGACGAGCCGT and three SEA-PHAGE genomes Citius, TPA4, and Vulture. He happens to know which of the three phage genomes this CRISPR spacer sequence is targeting (and thus where a match is expected), and he bets you a bag of donuts you can't figure out which one.
Download the example read and those genomes either by right-clicking the links above,
or with wget or curl, e.g.:
wget http://mcb112.org/w04/example_read.fa
wget http://mcb112.org/w04/Citius
wget http://mcb112.org/w04/TPA4
wget http://mcb112.org/w04/Vulture
(They are in FASTA format. You'll need a FASTA parsing function. You wrote one for pset 01.)
Search each genome with the GCAGCGGAGGAACCAGACATACTGACGAGCCGT read, finding the best score and position in each of the three genomes.
Again, computationally intensive. My scans took about 1-2 minutes per genome. I would have done this for more reads, but it would take too long. Python is slow! A C implementation of this would probably be at least 10x faster, and maybe 100x. Which genome did this read come from?
allowed modules
NumPy, Matplotlib, Pandas, Seaborn.
But you really only need NumPy for this.
You don't need to make any plots, but you can, so I included Matplotlib and Seaborn. You also don't need any Pandas data frames, but you can if you want to (like, in collecting a data table for the gap parameter sweep part).
import numpy as np
import matplotlib.pyplot as plt # optional
import pandas as pd # optional
import seaborn as sns # optional
%matplotlib inline
turning in your work
Submit your Jupyter notebook page (a .ipynb file) to
the course Canvas page under the Assignments
tab.
Name your
file <LastName><FirstName>_<psetnumber>.ipynb; for example,
mine would be EddySean_04.ipynb.
We grade your psets as if they're lab reports. The quality of your text explanations of what you're doing (and why) are just as important as getting your code to work. We want to see you discuss both your analysis code and your biological interpretations.
Don't change the names of any of the input files you download, because the TFs (who are grading your notebook files) have those files in their working directory. Your notebook won't run if you change the input file names.
Remember to include a watermark command at the end of your notebook,
so we know what module versions you used, in case we have to
reproduce some bug exactly. For example,
%load_ext watermark
%watermark -v -m -p numpy,matplotlib,seaborn,pandas,jupyter