Molecular Biology
A primer on Phage and Molecular Biology
Notes by Liana Merk (2024), adopted from Wendy Valencia-Montoya (2020), Irina Shlosman (2019), Kate Shulgina (2018), and Laura Bagamery (2016)
Preamble
Phage research has branched off in many directions, each of which has contributed in some measure to the edifice of molecular biology. One of the most notable directions was that of gene function and its regulation.
Salvador Luria, Nobel lecture, 1969
Many of the early discoveries in molecular biology arose from the study of bacteriophages, viruses that infect bacteria. For example, the Luria and Delbrück experiment showed that bacterial mutations conferring phage resistance occur spontaneously and are not induced by phage infection. They shared the 1969 Nobel Prize in Physiology or Medicine with Alfred Hershey, who -- with Martha Chase -- demonstrated that DNA is the genetic material. This upended the belief that proteins were the carriers of genetic information. Studying phage continues to be a powerful tool for understanding molecular biology. Throughout the course, it will be the stage in which we explore computational methods and inference fundamentals. In this primer, we intend to provide background to genomic information, conceptually and technically, and what kinds of biological questions genomes can answer.
The central dogma
All living organisms descent from a common ancestor and use the same molecules to store genetic information. However, every individual possesses a unique set of traits that distinguish it from members of other species and, in subtler ways, from other members of its own species. These traits, which are passed down from the organism's ancestors, are complex manifestations of units of heritable information known as genes.
How does an organism store, transmit, and interpret this genetic information that specifies its identity?
The central dogma of molecular biology posits that cells possess chemical codes detailing the assembly instructions for macromolecular machinery that promotes reactions underlying the fundamental physiological processes and states characteristic of the cell. Stored genetic material, in the form of DNA, is decoded to produce RNA intermediates, which are in turn decoded to build instruments of biological activity, proteins:
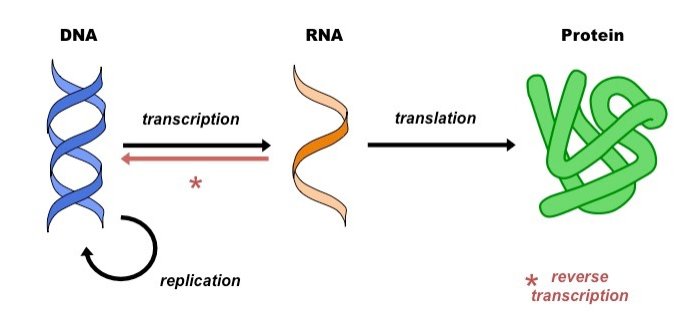
RNAs and proteins that are created in this way are referred to as gene products, and their synthesis is known as gene expression.
Genetic information at the molecular scale
On a molecular level, a gene is a sequence of deoxyribonucleic acid, or DNA. A molecule of DNA consists of two chains coiled around each other to form a double helix.

Each strand is built of nucleotide subunits, which are made of three parts: a five-carbon sugar (deoxyribose), a phosphate group, and a nitrogenous base. The four bases found in DNA nucleotides are known as adenine (A), guanine (G), thymine (T), and cytosine (C), and the sequence of these bases in DNA encodes the genetic information.
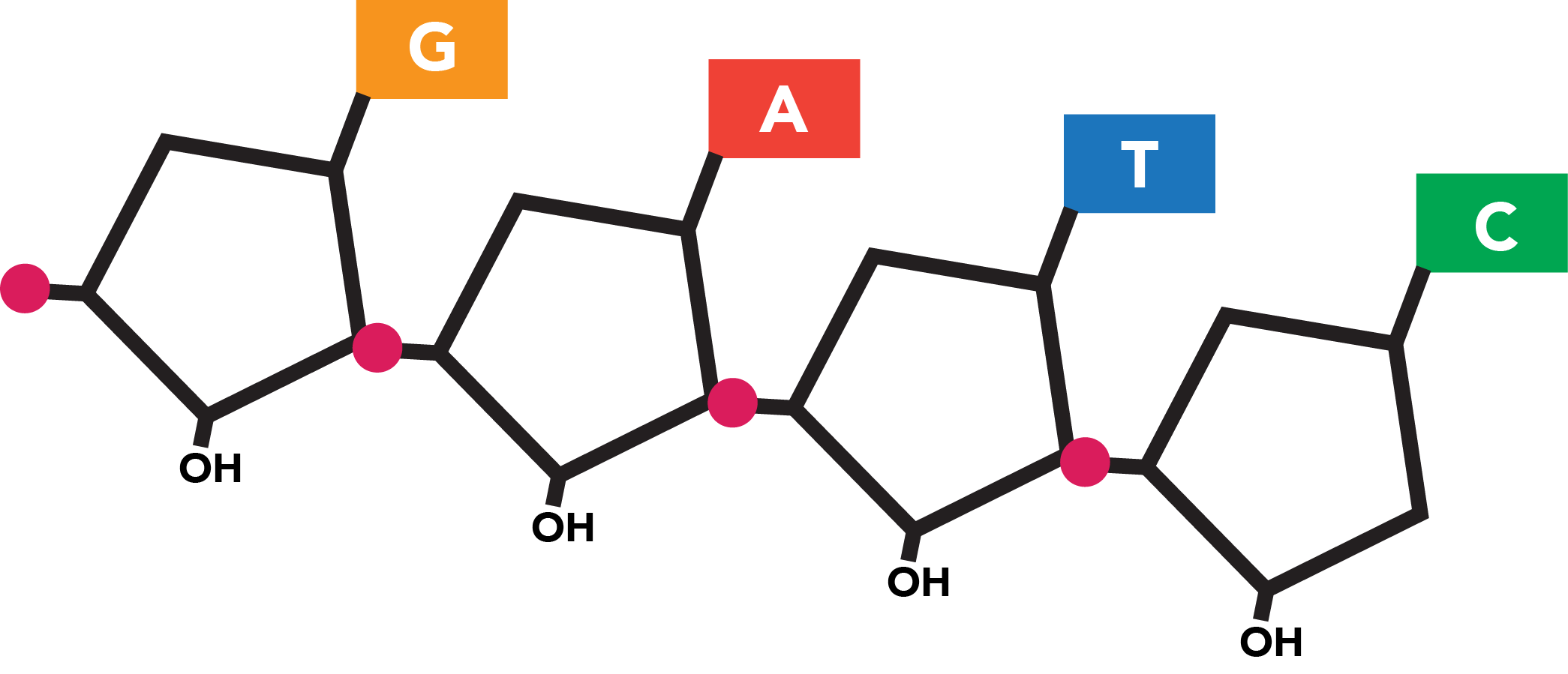
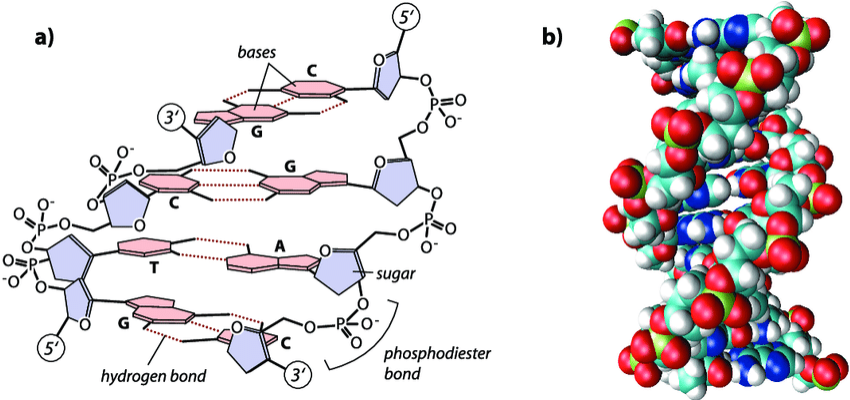
The double-stranded nature of DNA adds redundancy, as particular base pairs (bp) co-occur at the same position across sister strands and share weak bonds. These specifically pair A with T and G with C. The sequences of the two strands are thus complementary, with each reflecting the content of the other in a set of corresponding bases. This repetitive nature allows the fidelity of the sequence to be preserved in the event that one strand is damaged.
The information contained on each strand is also directional, as linkages within a DNA molecule imbue the structure with chemical polarity. Within the chain, a nucleotide is bound to the nucleotide that precedes it through its own phosphate group and to the nucleotide that follows it at its deoxyribose group. Under a particular convention for numbering the carbons within the five-carbon sugar, these linkages occur at the fifth carbon (which bears the phosphate group) and third carbon, respectively. Additional nucleotides can only be added to the end of the strand at which a nucleotide possesses an accessible third carbon. All reactions relying on the synthesis of new nucleic acid polymers--a category which includes both DNA replication and DNA sequence decoding (to be explained)-- must thus proceed along the polymer in a single direction known as 5' to 3' ("five-prime to three-prime"). This is the sole direction in which a strand "runs" and in which its genetic information can be read.
The two strands within a double helix are oriented such that the 5' end of one strand is paired with the 3' end of its sister. As the two strands run in opposite, or antiparallel directions, each sequence is, relative to its sister, a series of opposite bases running backwards, or the reverse complement.

Decoding DNA: transcription
Genetic information is extracted from DNA by transcription, in which the double helix unfurls and one strand serves as the template for the production of a molecule of ribonucleic acid, or RNA from the gene source. The structure of RNA is similar to that of DNA, but its phosphate-sugar backbone incorporates a slightly different sugar (ribose) and the base uracil (U) is substituted for thymine. Like thymine, uracil associates with adenine bases, although RNA is typically single-stranded and exhibits base pairing primarily in complex structures in which a single RNA strand partially folds against itself.
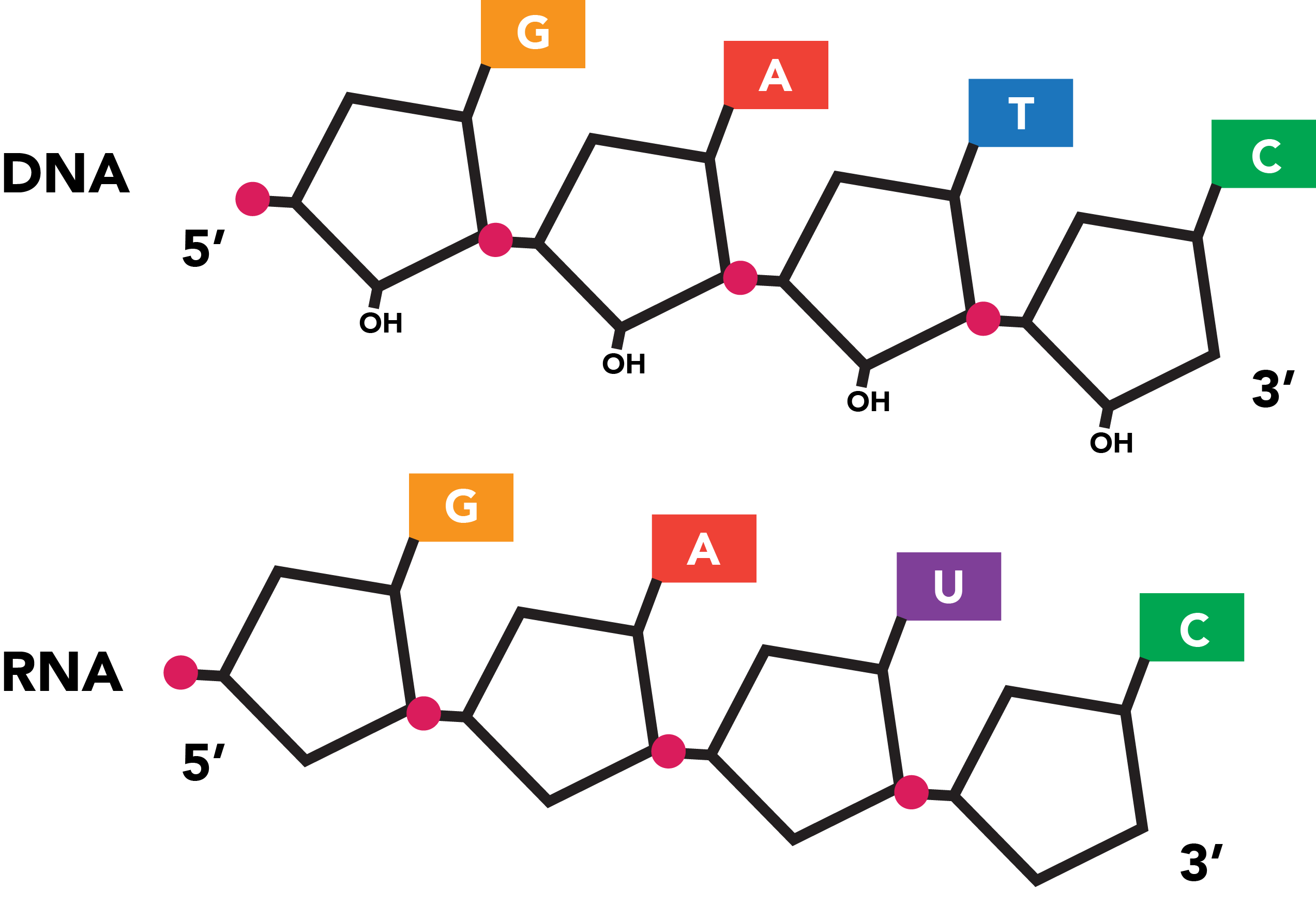
Transcription is capable of generating RNA products, known as transcripts, using either strand within a double helix as a template. The protein that catalyzes transcription, RNA polymerase unwinds a small portion of the double helix, which allows free RNA nucleotides to base pair to the template. The polymerase then links nucleotides into a continuous polymer by spurring the formation of phosphodiester bonds between the ribose sugars and phosphate groups of successive nucleotides. RNA, like DNA, can be synthesized solely from its 5' end along its 3' end, and this is the direction in which RNA polymerase moves down a nascent transcript.
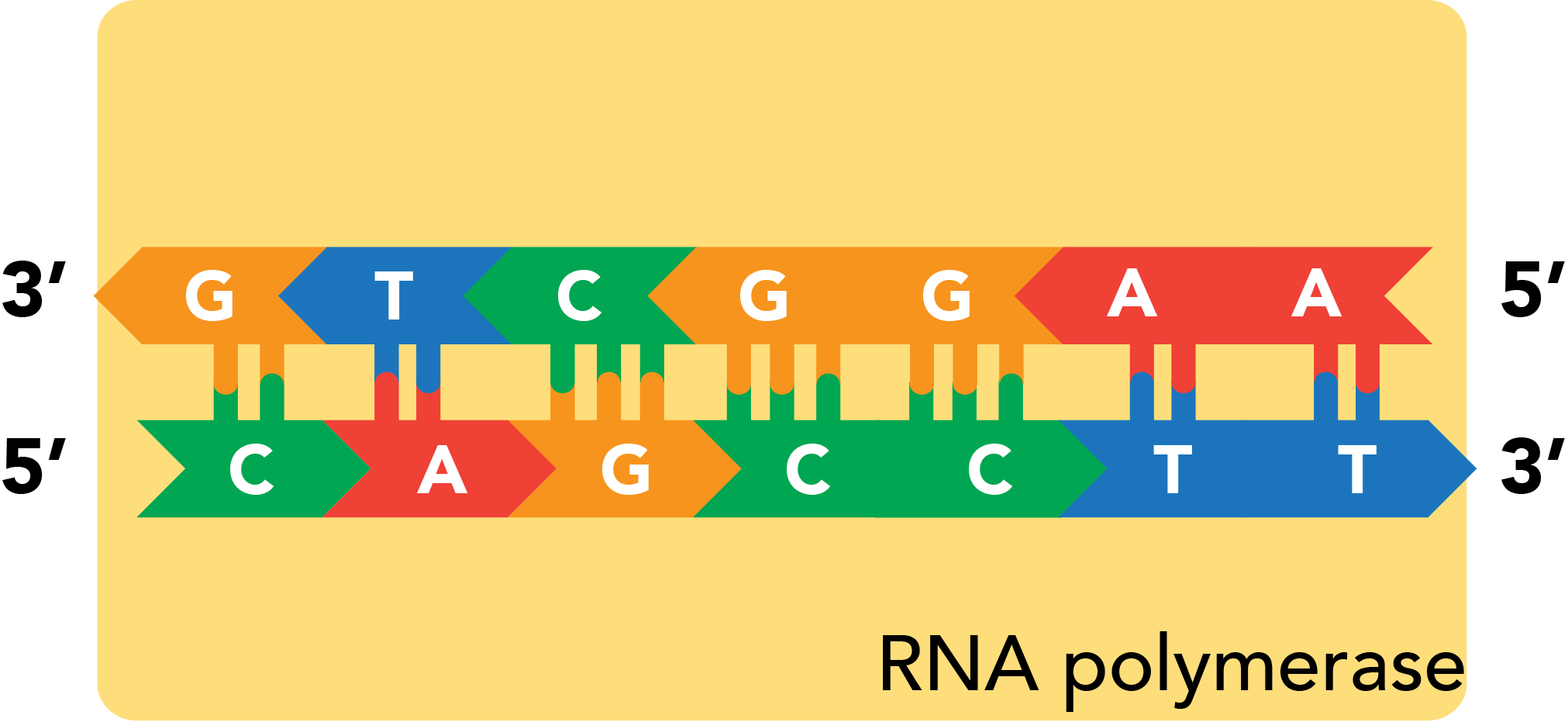



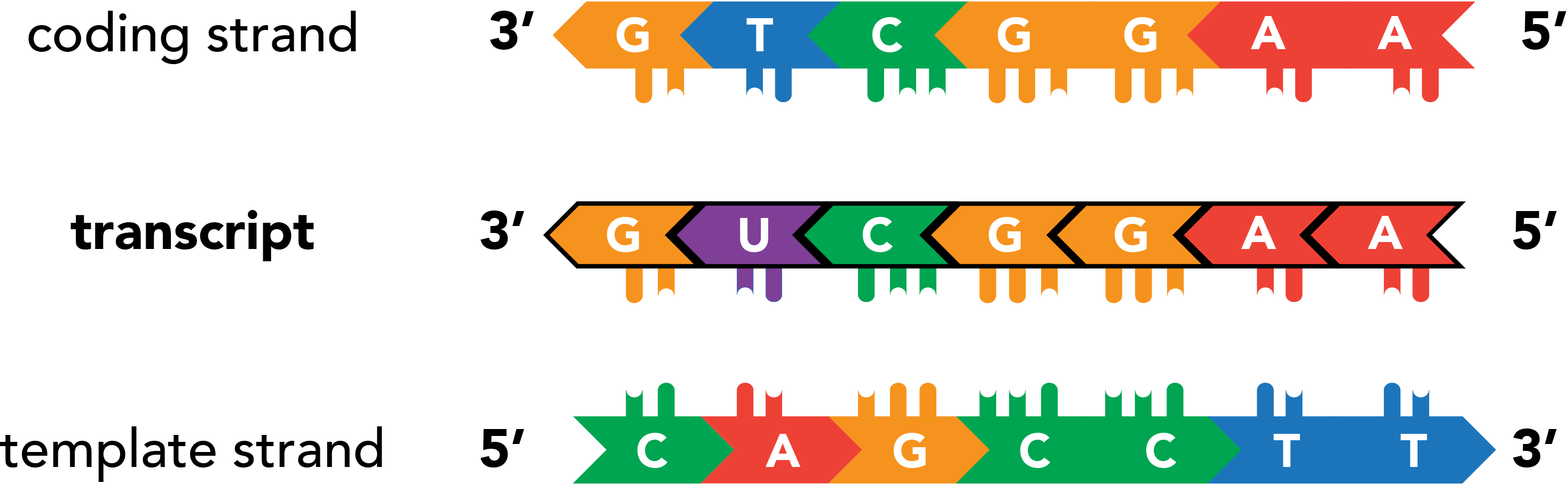
As the RNA lies antiparallel to its DNA source material, the sequence of a transcript will always be the reverse complement of its template. The template's sister strand, also the reverse complement of the template, features the sequence that will actually match the transcript (with U-T substitutions). This strand is accordingly known as the coding strand. Which strand is coding and which is template can vary among the many genes present within a stretch of DNA.
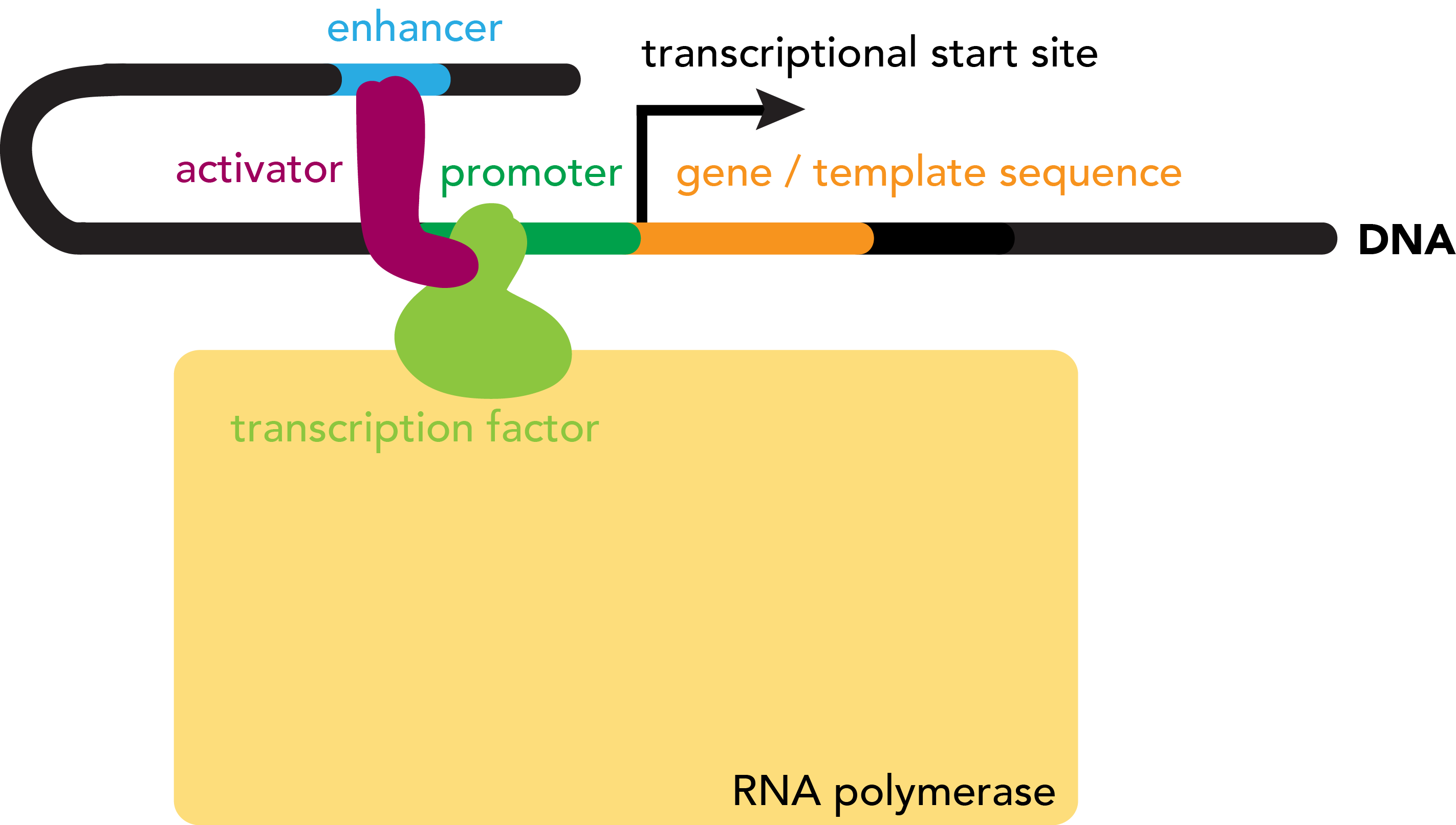
How does RNA polymerase know where to start transcribing a gene? A sequence that serves as template for an RNA molecule is prefaced by a promoter region, a stretch of DNA that defines the gene's start site. The promoter serves at the binding site at which RNA polymerase will be loaded onto the sequence prior to transcription.

Many factors influence the frequency with which RNA polymerase will bind to such a promoter region. These include the strength of the promoter--its intrinsic binding affinity for the polymerase--as well as the presence of absence of various accessory proteins, known as transcription factors, that influence the interaction between RNA polymerase and template DNA. Transcription factors may assist in either the recruitment or repulsion of RNA polymerase from a DNA sequence. Activators positively regulate RNA polymerase binding and thus transcription rates, while repressors negatively regulate these processes. Protein factors that alter the rate of transcription may also do so by binding DNA at regulatory regions other than the promoter, typically beyond the 5' end, or upstream of the gene but also past the 3' end (downstream), which can be relatively close to or far from the genes that they target. A complex interplay of all of these factors regulates, on the level of transcription, the extent to which a particular gene is expressed.
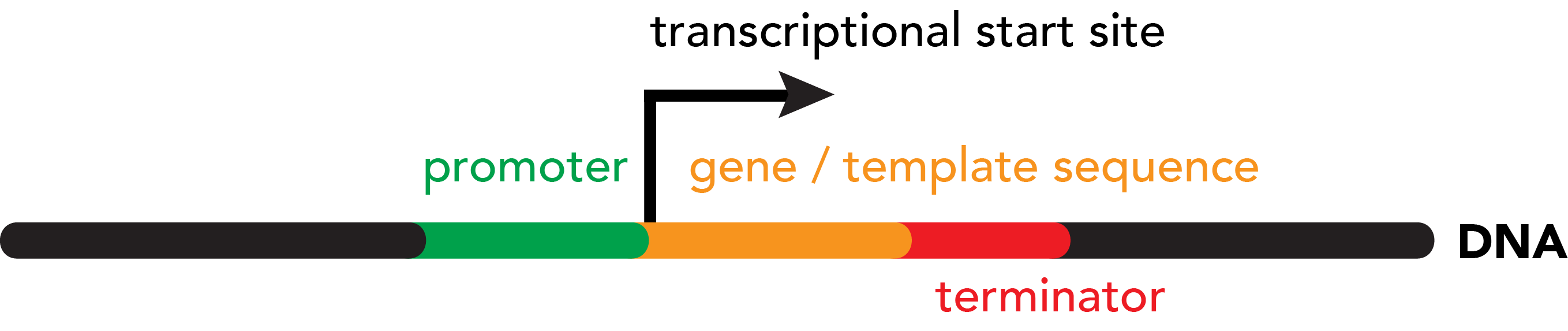
In contrast, the cues that mark the end of a gene are more straightforward. Directly downstream of the transcribed region, a terminator sequence triggers the release of RNA polymerase from DNA. This can occur in two main ways: rho-independent, or rho-dependent. Rho-independent terminators are characterized by the formation of a hairpin loop in the DNA, which stalls the RNA polymerase, eventually leading to it falling off of the template strand. Rho-dependent terminators involve a protein factor, rho, that binds to the RNA transcript and "chases" RNA polymerase. RNA polymerases slows down at the Rho-dependent terminator, allowing rho to catch up and physically knock the RNA polymerase off of the target DNA.
Decoding RNA: translation
In bacteria, even as RNA is still being transcribed, the 5' end of the nascent transcript is being translated into protein. This occurs by the ribosome, a complex of proteins and ribosomal RNA (rRNA) that catalyzes the synthesis of proteins from RNA templates.
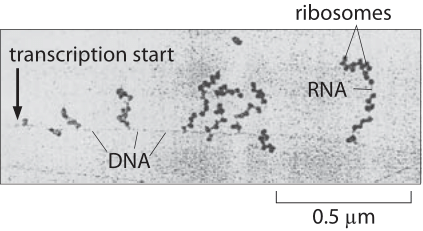
The RNA transcript serves as the template for the production of a protein, which consists of one or more polypeptide chains. A polypeptide chain is a linear sequence of amino acid subunits. Twenty different amino acids can appear in polypeptides, yet they are specified from a nucleotide code comprised of just four distinct base types. This level of variation is possible by specifying amino acids with groups of three consecutive nucleotides. The set correspondence between RNA triplets, or codons, and their resulting amino acids is the genetic code.
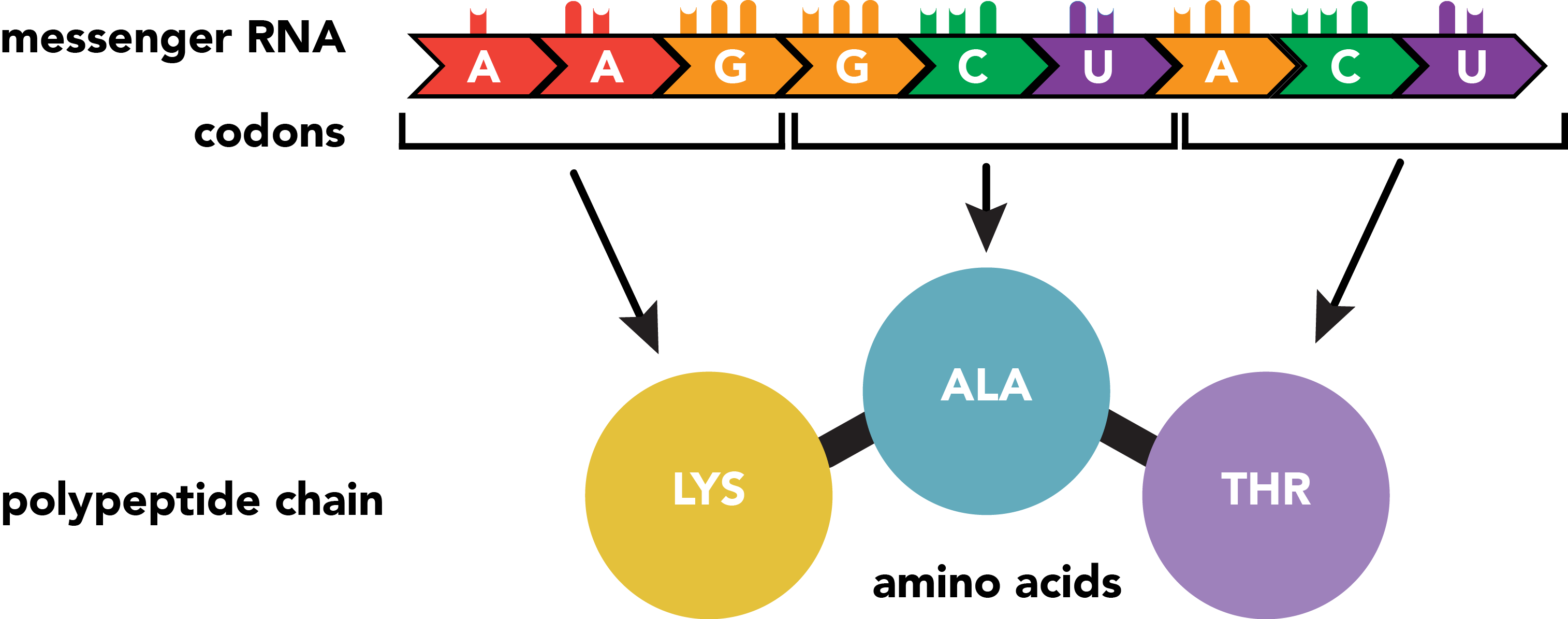
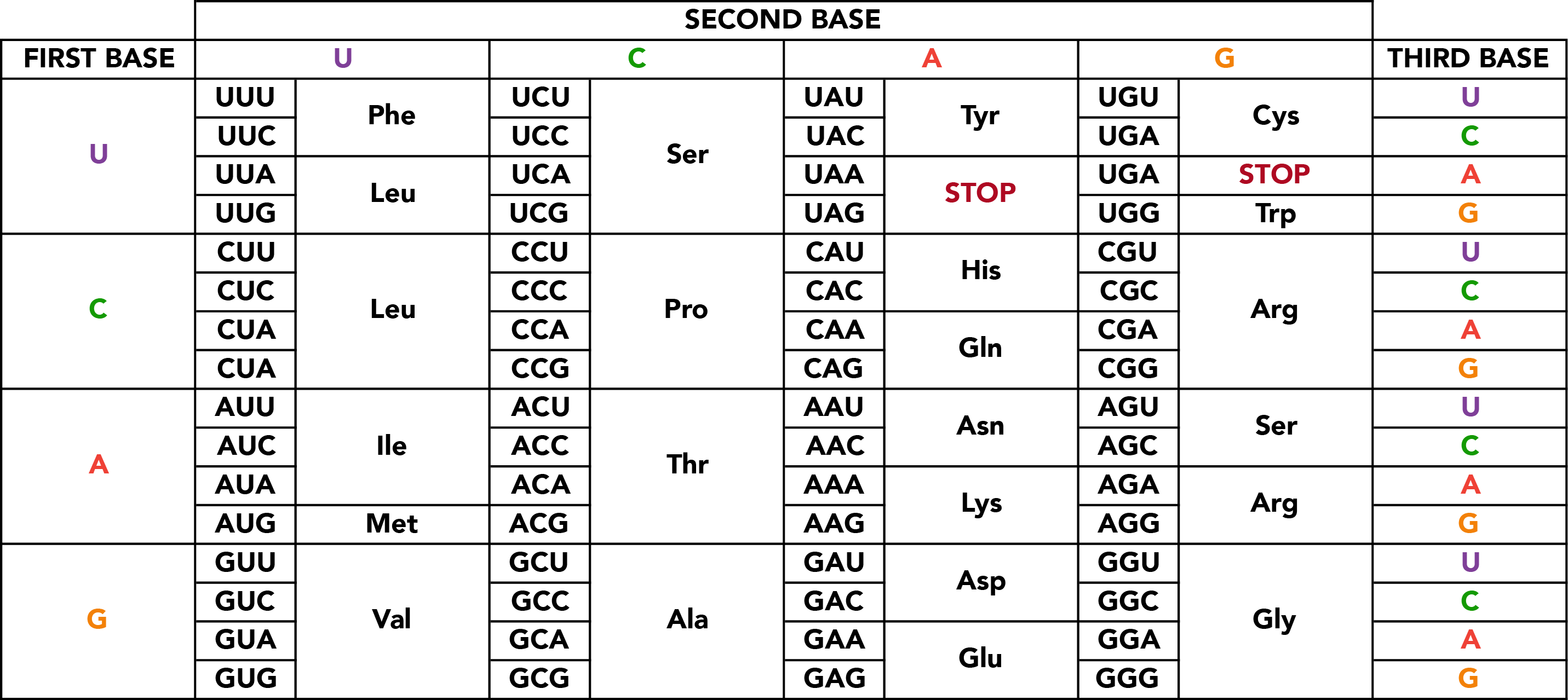
Of the sixty-four possible nucleotide triplets, the majority specify particular amino acids with redundancy in codons, but one encodes both an amino acid (methionine) and a general signal to initiate polypeptide synthesis if it has not already begun. This start site is not necessarily the first codon within the mature mRNA, and sequence upstream of it, the 5' untranslated region (UTR), will not be integrated into the polypeptide chain. Similarly, translation ceases with one of the three stop codons which specify not amino acids, but the cessation of translation itself. Sequence further downstream, the 3' UTR is also omitted from the protein sequence. These sequences perform regulatory functions in recruiting translational machinery and otherwise regulating translation rate, mRNA localization and stability, and similar properties.

The act of translation involves three distinct classes of RNA molecules: the mRNAs that serve as coding templates; transfer RNAs (tRNAs), which act as adaptor molecules that bind particular amino acids and deliver them to the site of the appropriate codon; and ribosomal RNAs (rRNAs), which function as critical subunits of ribosomes, the sprawling cellular machines that perform the synthetic activity of attaching an amino acid to a nascent polypeptide chain.
The ribosome contains three sites that bind mRNAs and tRNAs. The A site binds aminoacyl-tRNAs, or charged tRNAs, bound to their amino acid cargo. The P site binds peptidyl-tRNA, an RNA molecule that carries not only its own target amino acid but the entire nascent peptide chain, to which it is bound through the last added amino acid residue (the tRNA's own target). The E site is where tRNAs with cargo that has been successfully integrated into the polypeptide exit the ribosome.
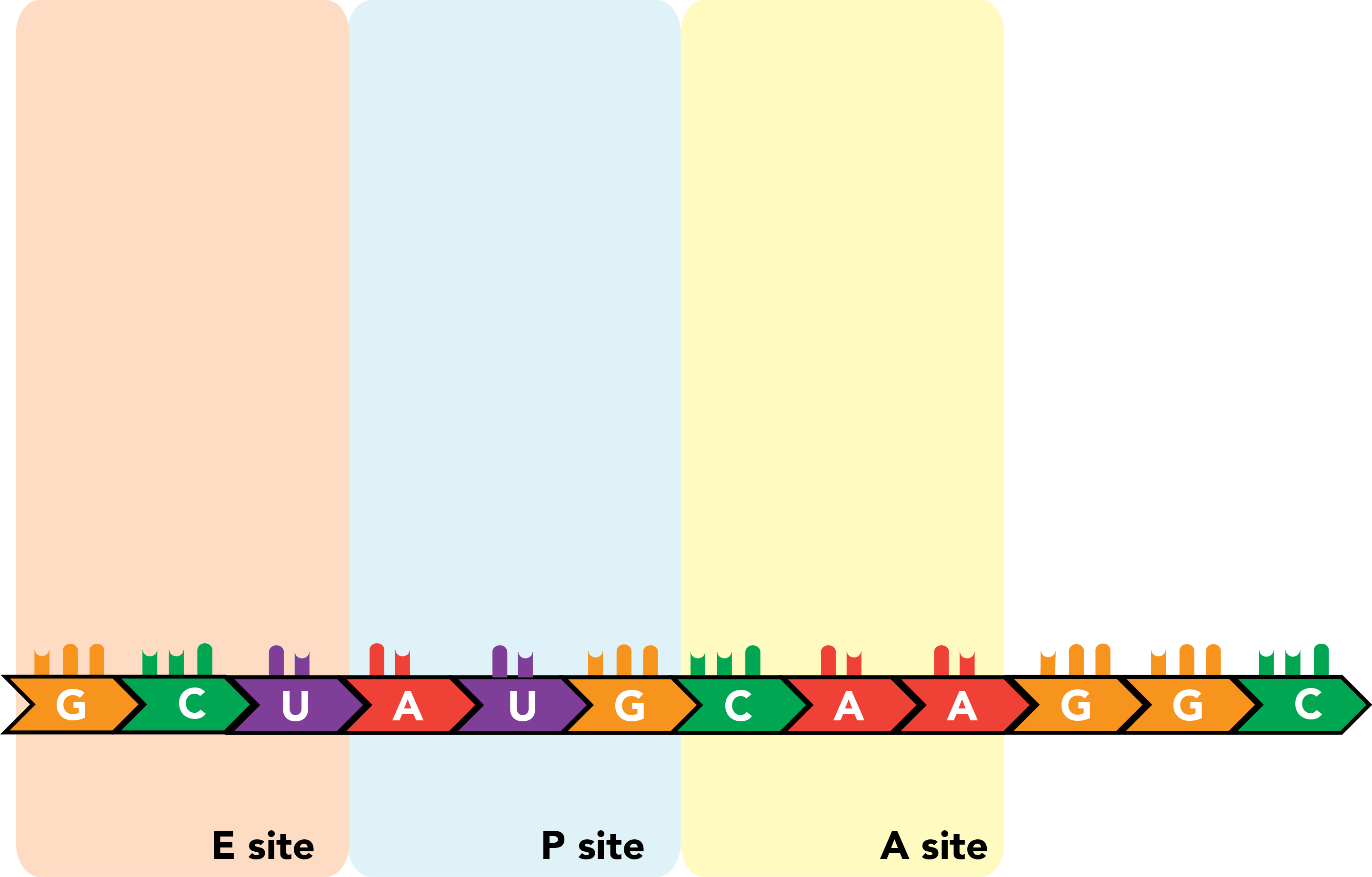
Translation is initiated with a start codon (AUG) in the P site of a partially assembled ribosome. An appropriately charged tRNA is folded into a confirmation that permits binding of an amino acid (here, methionine) at a single-stranded stretch at its 3' end and leaves an anticodon, the reverse complement of the mRNA codon specifying the same amino acid, exposed along another face of the molecule. For the first translated peptide, this tRNA is an initiator tRNA, and its binding to the start codon will promote full ribosome assembly.
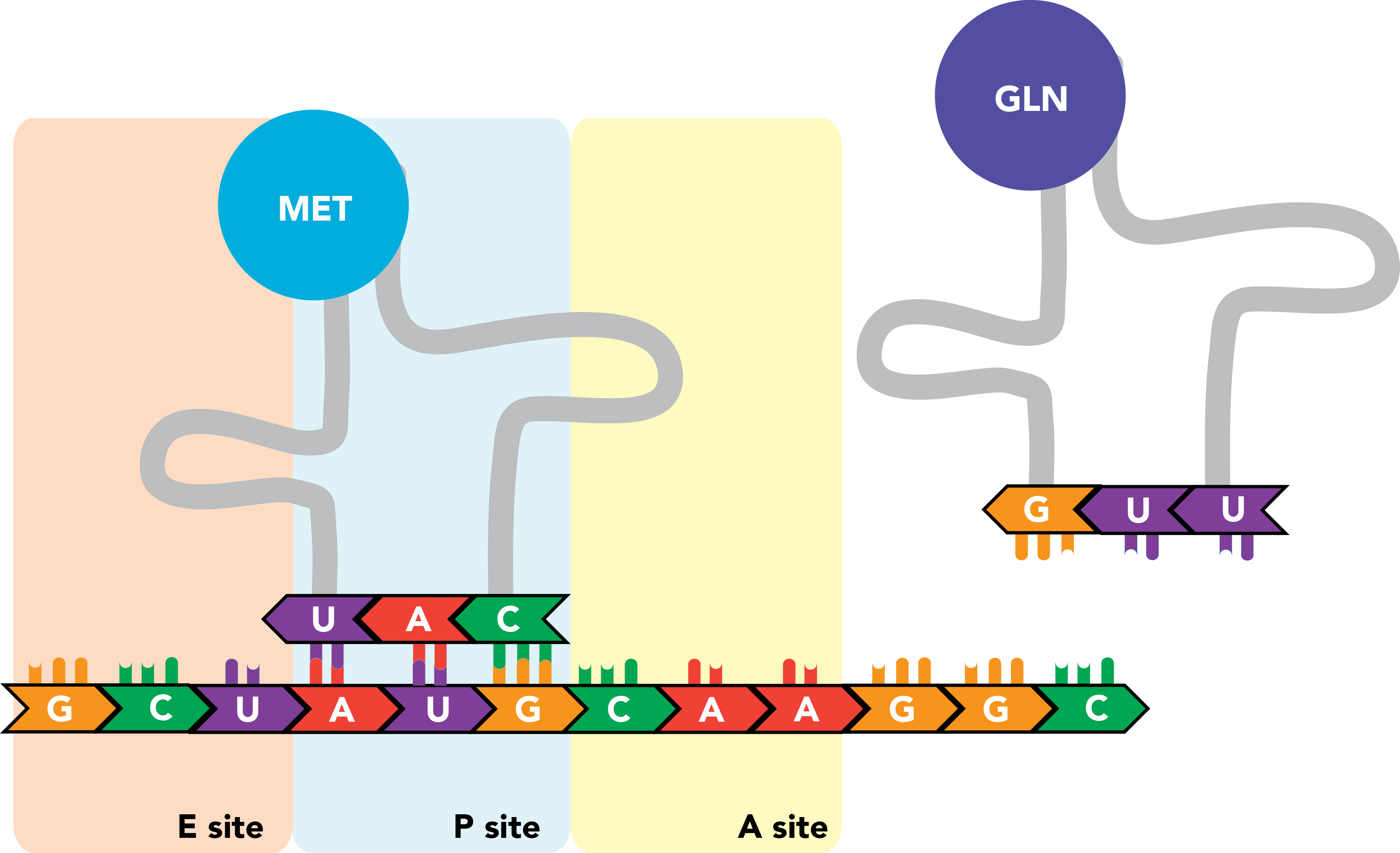
Once this occurs, a tRNA complementary to the next codon, which resides in the A site, enters the ribosome, and the codon and anticodon engage in base pairing. There are now amino acids in two ribosomal compartments: one attached to the initiator tRNA in the P site, and the next successive amino acid attached to the tRNA in the A site.
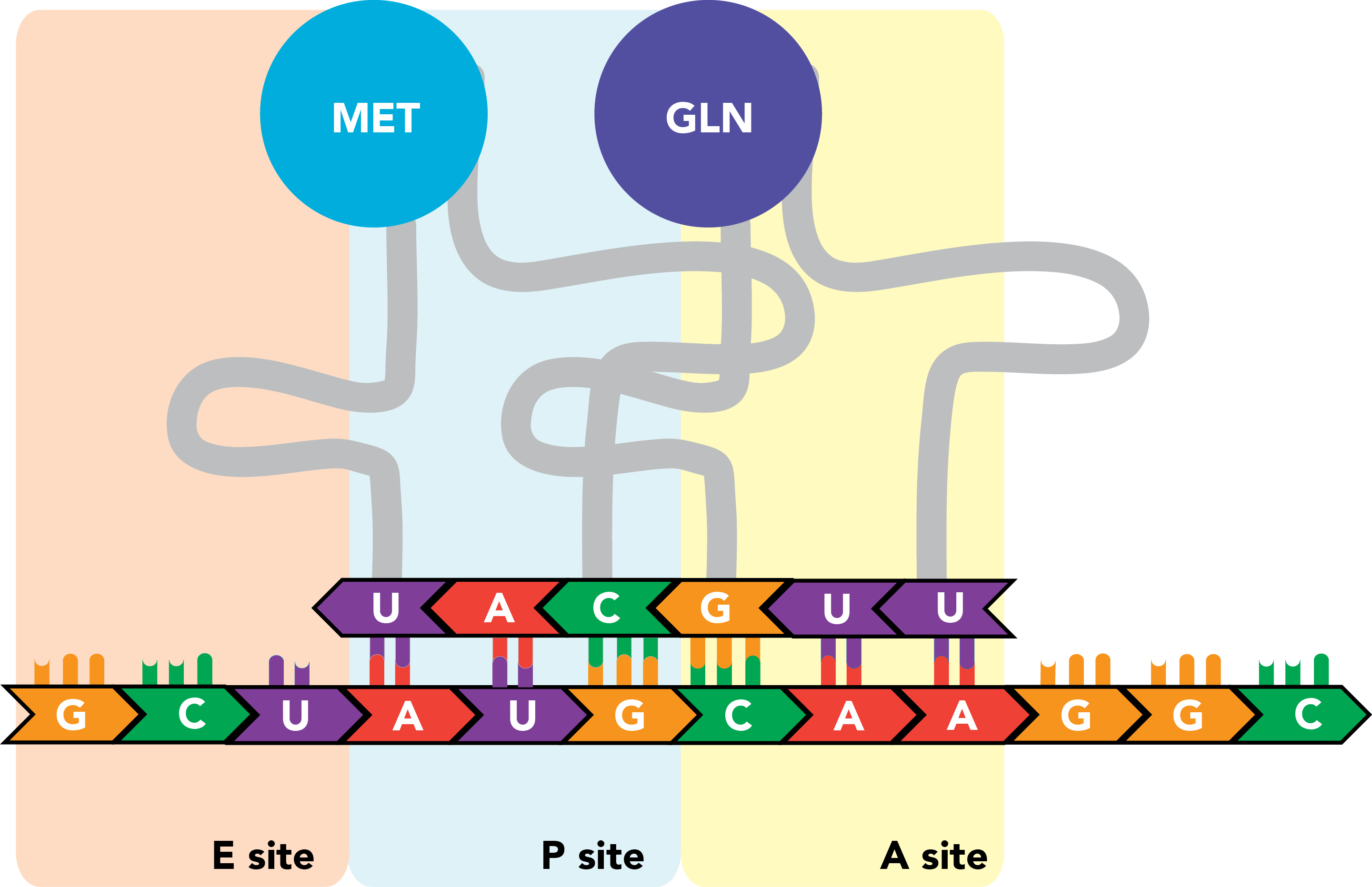
The peptide in the P site is transferred from its tRNA onto the amino acid in the A site, and the two are linked by a polypeptide bond.
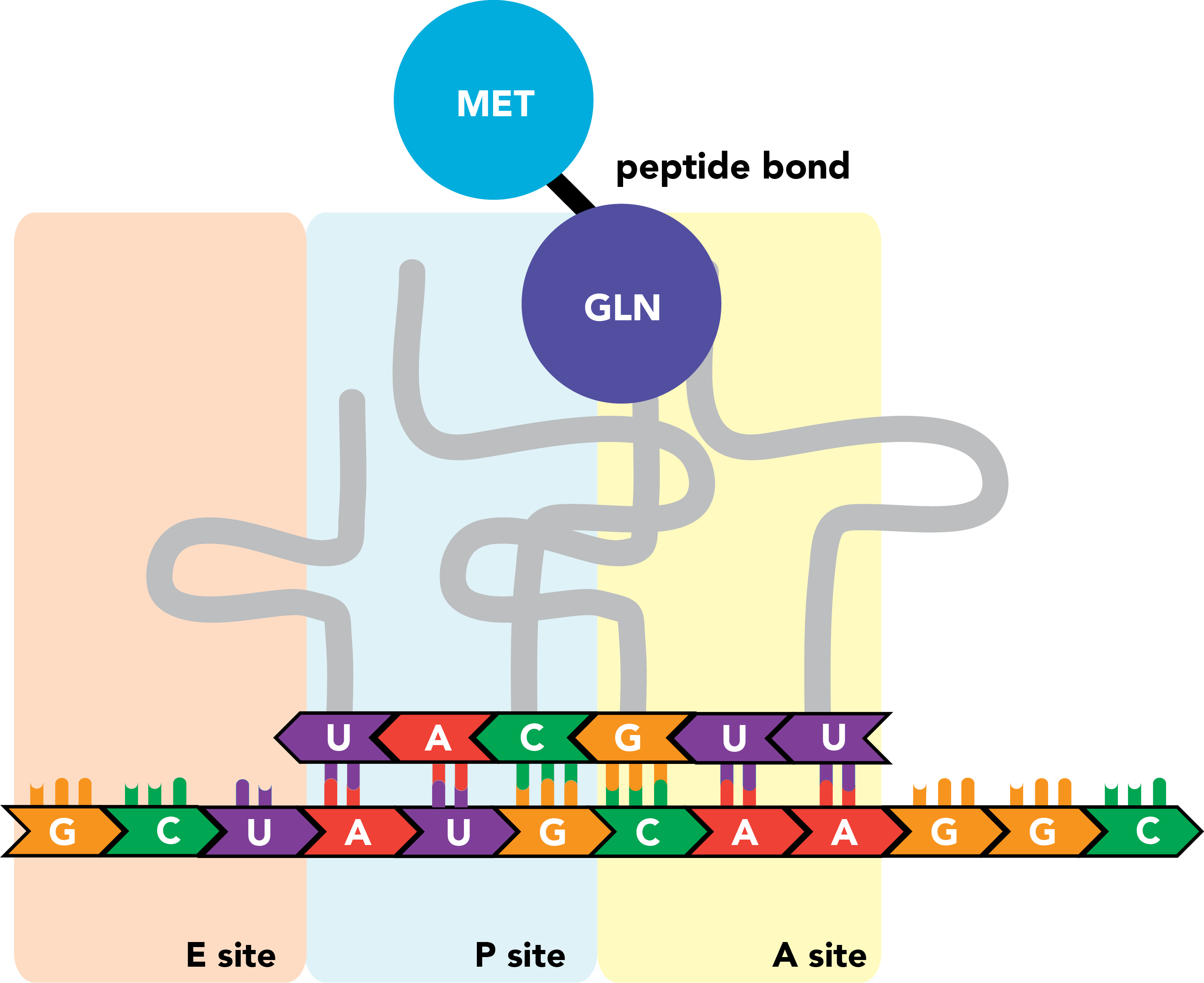
The ribosome then shifts once again.
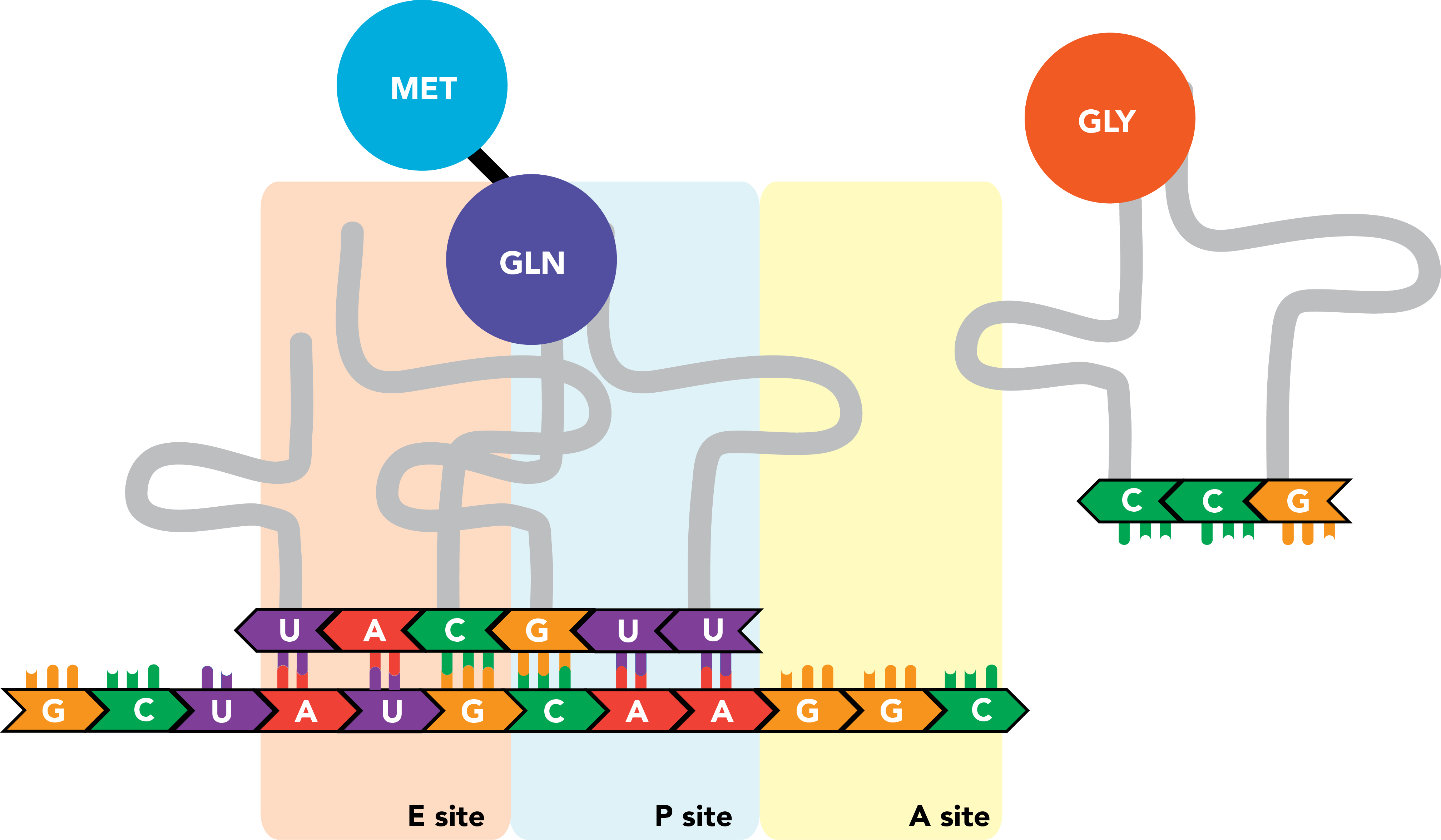
The initiator tRNA, now in the E site and relieved of its cargo, is released from the ribosome and the transcript. Meanwhile new tRNA binds to the next successive codon in the A site.
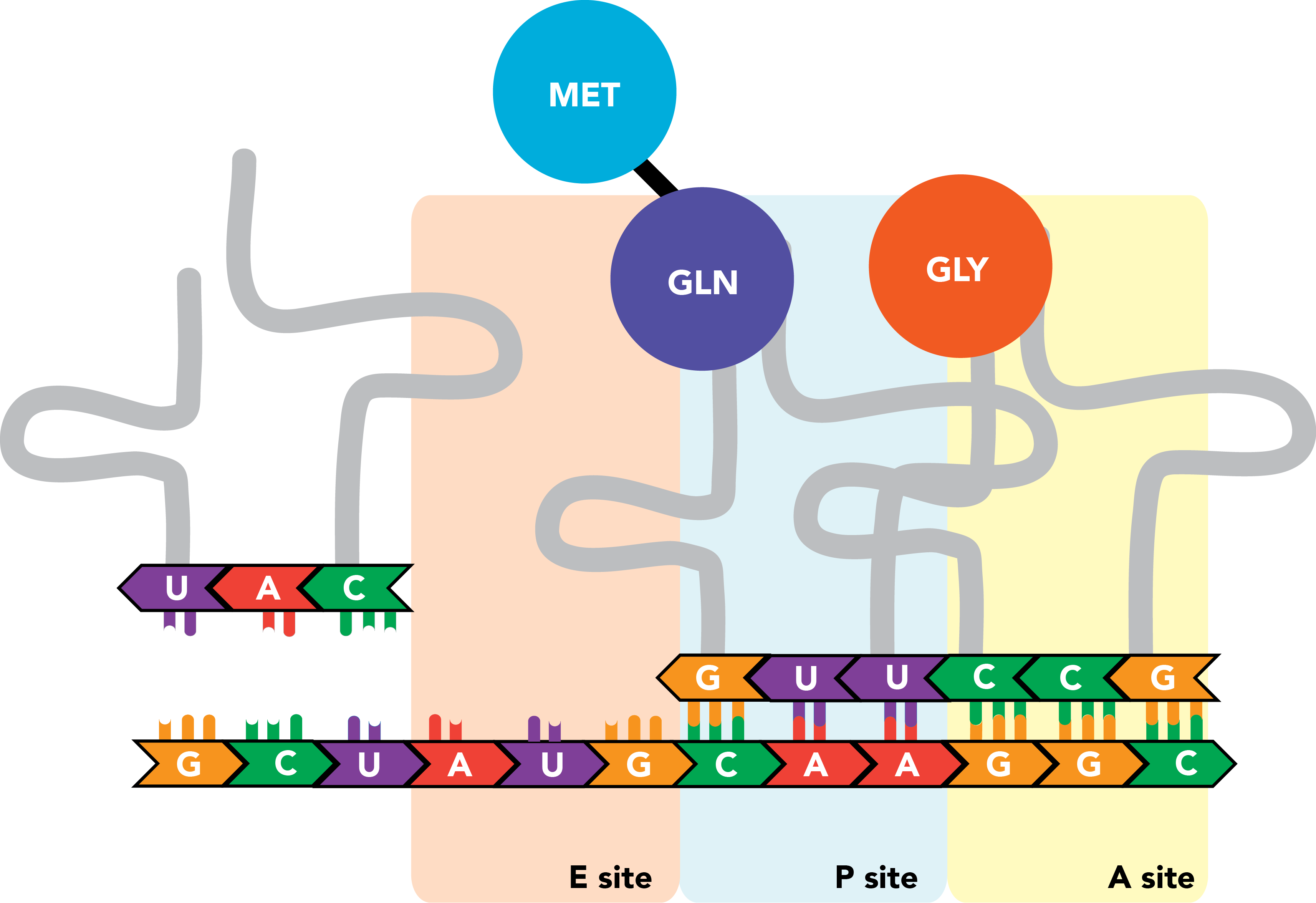
The nascent polypeptide is transferred to the end of this next amino acid.
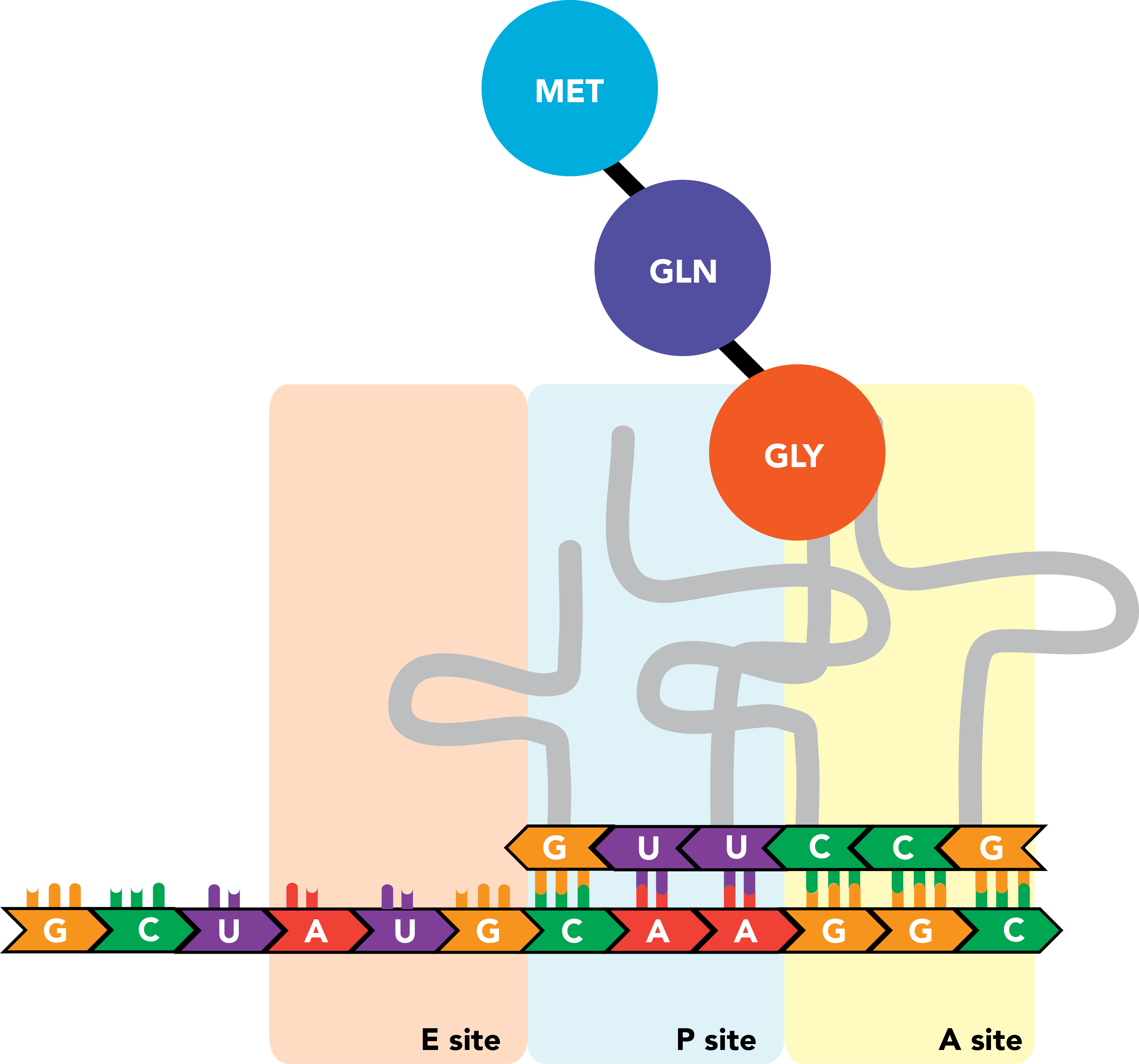
This process repeats itself as the ribosome moves processively along the mRNA molecule, triplet by triplet, from the 5' to 3' ends of the transcript, until a stop codon appears in the A site. There, release factors bind the codon and prompt the ribosome to eject its completed polypeptide chain and dissociate from the mRNA.
Finally, one or more linear chains of polypeptides fold into three-dimensional structures to form proteins. These proteins perform critical functions in virtually all biochemical processes within a cell--including, among many others, the transcriptional and translational activities described above.
DNA sequencing
To understand DNA sequencing, it is necessary to understand the mechanics of DNA replication. As in transcription, DNA replication begins with the unwinding of the double helix (the denaturation of the double-stranded macromolecule) to produce single strands that function as template. Free nucleotides are added sequentially to the growing polymer as they bind to their complementary base pairs on the template. This synthesis occurs exclusively at the 3' end of the growing chain. The result is a DNA strand that, as in the double helix, is the reverse complement of its template, with the two strands in antiparallel orientation. During replication of the genome, both parental strands serve as template simultaneously.
Methods for identifying the order of nucleotides within a sequence typically do so by exploiting the processive nature of replication, using the query DNA as template and detecting the addition of particular nucleotides with base-specific inhibitors and/or fluorescent dyes. The oldest (and still quite common) technique is Sanger sequencing. In it, source DNA is divided among four reaction tubes. Each tube contains the same short DNA sequence that is the reverse complement of a particular site on one of the source strands, where it will bind and seed polymerization of a new strand, directing replication and thus sequencing data toward this starting point. This sequence is known as a oligonucleotide, or primer. Each reaction also contains the protein that incorporates nucleotides into DNA (DNA polymerase) and DNA nucleotides (deoxynucleotides, or dNTPs---this is the generalized term for nucleotides of all four bases: dATP, dGTP, dTTP, and dCTP. The reactions differ in that each contains a dideoxynucleotide (ddNTP) with one of the four bases. This nucleotide is incorporated into nascent DNA normally, but because it lacks the 3' hydroxyl group that allows for the addition of additional nucleotides at the 3' end of a growing strand, it will terminate the sequencing reaction and serve as the 3' terminal end of the chain. The four ddNTPs are present at low concentrations relative to the dNTPs, so that when replication requires the relevant base, the terminating ddNTP is incorporated with low probability, and synthesis continues with the proper dNTP in the majority of occurrences.
When multiple copies of the template are replicated under these conditions, the result is a series of DNA fragments of different sizes, all of which incorporated a ddNTP at a different instance of the appropriate base and terminated afterwards. The lengths of these fragments can be used to infer the location in the sequence at which the base is present, and with four reactions--one for each base--the identity of the base at every position in the source DNA can be determined. In the original Sanger method, ddNTPs are labeled radioactively. The DNA fragments are injected into a meshwork (an agarose gel) and an electric field is applied, causing the negatively-charged phosphate backbones of DNA molecules to propel them down the gel. With the lattice structure of the gel impeding larger molecules, the rate of migration through the gel is dependent on the size, so that fragments are separated based on length. Autoradiographic detection techniques on the sorted gel can then detect the exact lengths of the hot fragments, revealing precisely where in the sequence a ddNTP was incorporated as an indicator of nucleotide identity at that site. In the modern adaptation, each ddNTP is labeled with a fluorescent dye of a different color, allowing the incorporation of each ddNTP to be distinguished within a single reaction instead of four.
The Sanger method remains a simple and reliable way to perform sequencing with high fidelity, although the sequence quality eventually deteriorates, limiting the interval of DNA that can be sequenced in a single reaction. This is typically a sequence fragment, or read, of several hundred base pairs. When determining the sequence of longer stretches of DNA, multiple Sanger reactions must be performed with primers spaced across the region to cover its sequence collectively.
Sanger sequencing represents the first generation of sequencing technology; the high-throughput permutations that have arrived since are known as next-generation sequencing. These are capable of performing sequencing from multiple sites in parallel to sequence full genomes with efficiency.
The vast majority of high-throughput sequencing now relies on a next-generation sequencing technique called Illumina sequencing (again, corporate branding). Illumina sequencing begins with the random fragmentation of larger DNA sequences into short fragments, of about 100-500 nucleotides depending on the instrument. Adaptor sequences that are serve as unique barcodes for each fragment are ligated onto both fragment ends. Such a fragment collection is known as a library. The contents of the library are denatured and flown over a plate covered with small sequences that are complementary to the adaptors. Similar to Sanger sequencing, the plate is exposed to reaction mixture containing primer, DNA polymerase, and four distinctly-labeled fluorescent nucleotides, but in Illumina sequencing, all of the nucleotides added inhibit DNA replication once they are incorporated--but reversibly so, as each consists of a nucleotide with a removable inhibitor attached its 3' end. In the presence of this reaction mixture, a single nucleotide is added, and replication is inhibited thereafter. The identity of this nucleotide is determined from its fluorescence. Then the reaction mixture is washed out the inhibitors are cleaved from the nascent DNA chains, and fresh reaction mixture is added. This continues for many, many cycles.
The short read lengths characteristic of Illumina sequencing mean that sequence data is heavily fragmented and reads must be compiled into longer, more coherent forms in post-processing. (This complex, randomly fragmented output is also why such sequencing techniques are sometimes referred to as shotgun sequencing.) To reconstruct a genome, low-quality and extremely fragments are purged and the remaining fragments are clustered based on matching, overlapping sequence information, forming contigs (continguous sequences). When the genome of the organism has previously been characterized, contigs can be mapped, or aligned, directly to this reference genome by scanning the genome for a matching sequence. When there is no prior sequence information available, the fragments must be assembled de novo. In this case, contigs are stitched together into a final assembly without the benefit of a reference template. Genomic data can then be annotated, to mark features of interest, such as genes, transcriptional binding sites (and many more) within particular base pair regions.
One technique that has greatly aided in the assembly of genomes de novo is long-read sequencing. Specifically, Nanopore and PacBio sequencing are capable of reading molecules tens of kilobases at a time, albeit at lower throughput. Nanopore sequencing relies on passing DNA through a small pore, or nanopore, and measuring how the electrical current changes. The identity of the bases can be inferred from the current, even as the DNA is being passed through the pore. This method is capable of producing reads that are tens of thousands of base pairs long, which can be used to assemble genomes with fewer contigs and gaps. PacBio, on the other hand, creates a circular DNA strand that is sequenced using fluorescent nucleotides, similar to Illumina sequencing. However, a single DNA molecule is circularized and sequenced many times in a method called "circular consensus sequencing". This allows for decreased error rate, and the ability to detect larger structural variants or repetitive regions in a genome.
When you look at a genbank or fasta file, you may notice information about the sequencing technique in the header. Keep your eye out for the terms we've just learned!
How to build a phage
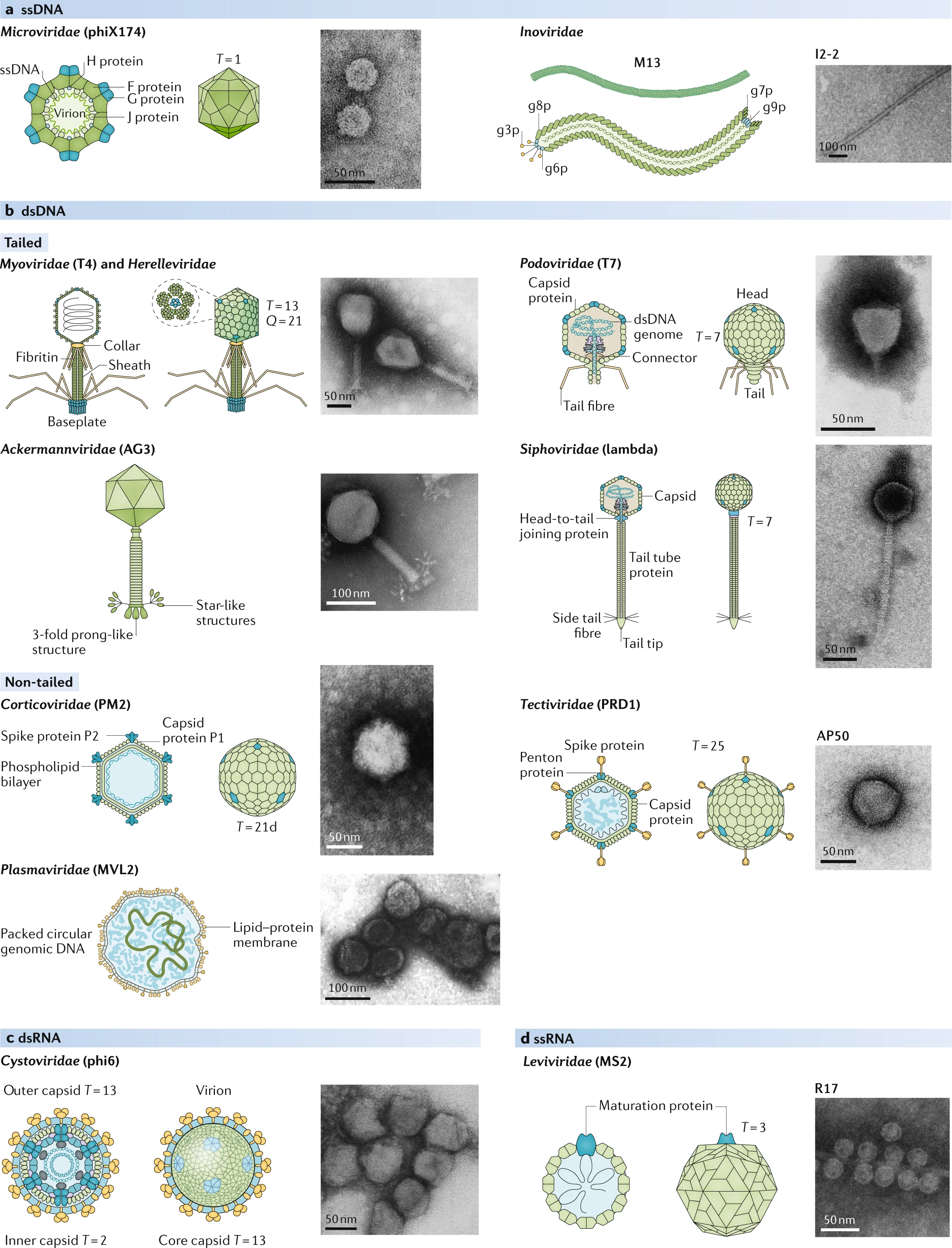
Phage have adapted to survive in nearly every environment we have looked in, from the human gut to boiling hot springs. They are incredibly diverse, and their genomes reflect this. Phage genomes can be composed of DNA or RNA, single stranded or double stranded, ranging from around 5,000 to 800,000 base pairs. While the canonical image of phage T4 may look familar, there exists a range of phage morphologies:
As we learned in lecture, phage genomes are very tightly packed, meaning the coding density is extremely high. This is because phages are under selective pressure to be as efficient as possible, packaging only the essentials, and having very little tolerance for extraneous DNA. Because of this, we can -- by eye -- determine a function for almost every gene. Let's take a look at phage lambda, a well-studied phage that infects E. coli. The lambda genome is around 48,000 base pairs long, and contains 73 genes.
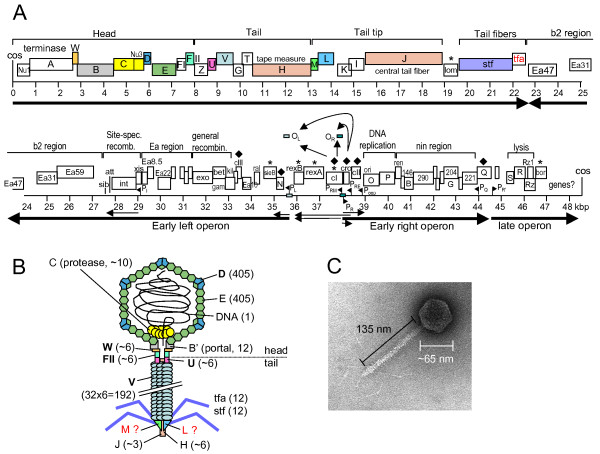
Lambda is a very interesting phage that can exist in the lysogenic lifestyle, which means it can integrate into a bacterial genome, lying dormant until some stressor activates it to enter the lytic lifestyle. This is mediated by a repressor, called CI, which binds to the lambda genome at a site an "operator". This regulates the Pr and Pl promoters, tamping down their expression of the lytic genes involving replication and recombination. However, when the bacterial cell undergoes some kind of stress (usually DNA damage), CI is degraded, and the lytic genes are expressed. This causes the production of more Lambda genomes, and subseqeuntly the structural proteins needed to encapsulate new virions. The late operon also contains lysis genes, which eventually pop the (former) bacterial host, and around 100 Lambda virions are released.
We can also explore the genome in an interactive way, through a couple of tools:
-
Phagescope genome viewer tool. Search for a new phage here
Feel free to play around with these, and other phage genomes (some favorites are an inovirus: AB334721, T4: NC_000866, and a microvirus: NC_002643) to get a sense for the types of genes that are present, as well as their organization. We will learn more about how to analyze these genomes in the coming weeks.
Further reading
If you are interested to learn more about Lambda phage and lysogeny: A Genetic Switch
If you find the origins of our field interesting and would like to learn more: Phage and the origins of molecular biology